Explanation for the choice of comparators
To address the neurohormonal response associated with subclinical hemodynamic deterioration, dobutamine, an inotropic agent, was selected. Dobutamine increases cardiac contractility and reduces systemic vascular resistance, resulting in a decreased left ventricular afterload [15]. In dogs with experimentally induced low cardiac contractility, reduced cardiac output, and hypotension, dobutamine administration resulted in dose-dependent increase in cardiac contractility as well as output, effectively restoring arterial blood pressure [18].
The standard dosage for acute heart failure and CS typically involves an initial dose of 2–3 μg/kg/min, which can be titrated up to 20 μg/kg/min. For this trial, a low maintenance dose of 5 μg/kg/min was selected to mitigate the potential risk of tachyarrhythmias occurring in the setting of AMI [18]. No differentiation was made regarding patients treated prior to admission with beta-blockers. Furthermore, a low dose of dobutamine was chosen to minimize cardiac energy consumption and to achieve a balance of vasoconstriction and vasodilation [19].
Intervention description
Patients will receive a 24-h IV infusion of 5 μg of dobutamine per kilogram of body weight per minute or placebo (saline). The infusion is administered as a weight-standardized preparation, mixed in isotonic saline at a rate of 5 mL per h, resulting in a total volume of 120 mL.
Individualized infusions for each patient are prepared by a collaborating cardiac unit at the center, ensuring maintenance of blinding for the treating and administering unit, including clinical staff, patients, investigators, and sponsors. Each infusion kit is labeled for safe and accurate administration. The assigned treatment arm will remain visually indistinguishable. Infusions will be administered through either a central or peripheral IV line using standard infusion pumps. The administration will be initiated as early as possible after obtaining informed consent and no later than 2 h after the PCI procedure.
Criteria for discontinuing or modifying allocated interventions
Patients retain the right to withdraw from the study at any time and for any reason, without any consequence for the future medical care, in compliance with relevant legislation and ethical guidelines. If a patient decides to withdraw their consent to participate, the administration of the study treatment will be immediately discontinued. Data collected up to the date of withdrawal will be kept.
In certain clinical situations, the sponsor and/or the investigator may decide to withdraw a patient from the study. This decision can be made if there is a significant intercurrent illness or if the investigator or sponsor determines that termination is in the patient’s best medical interest.
Patients who withdraw may still be offered the option to continue sampling after individual agreement. An end-of-study case report form will be completed to document the patient’s final termination in the study, including an explanation for the withdrawal.
The dobutamine/placebo infusion rate may be reduced as per the protocol to 2.5 μg/kg/min (half dose) if persistent tachyarrhythmia occurs (ventricular rate ≥ 130/min for > 30 min). Additionally, the treating physician has the discretion to modify or halt the dobutamine infusion if deemed necessary. Any such changes will be recorded.
Strategies to improve adherence to interventions
Detailed standard operating procedures (SOPs) are used to standardize procedures, and clinical personnel receive role-specific training. To ensure access to support, an investigator is available 24/7. The intervention is administered only once to minimize errors. Dose modifications are documented alongside clinical observations for safety on a case report form.
Relevant concomitant care permitted or prohibited during the trial
Screening runs concurrently with acute patient treatment. The trial intervention supplements current standard guidelines for treatment of AMI patients [20, 21]. No delay in treatment is expected, and no restrictions apply to concomitant care or interventions. All interventions before or during admission are documented for subsequent analysis and reporting.
Provisions for posttrial care
Patients in the study are covered by the Danish Patient Compensation Scheme, like other healthcare recipients under the Danish Patient Compensation Act. After the final follow-up visit, any observed clinical or patient-reported indicators of new or worsening diseases (including mental health and depression) not currently under diagnosis or treatment will be assessed and referred if necessary.
Outcomes Primary endpoint
The primary endpoint is the between-group difference (dobutamine/placebo) in peak proBNP within 48 h from randomization.
Secondary endpoints
Between-group differences (dobutamine/placebo) in acute and follow-up hemodynamic function evaluated by TTE measurements
Between-group differences (dobutamine/placebo) in pulse rate and blood pressure within 48 h from infusion
Between-group differences (dobutamine/placebo) in proBNP at three-month follow-up
Between-group differences (dobutamine/placebo) in troponin T, lactate, and CK-MB before infusion, 12, 24, 36, and 48 h after infusion
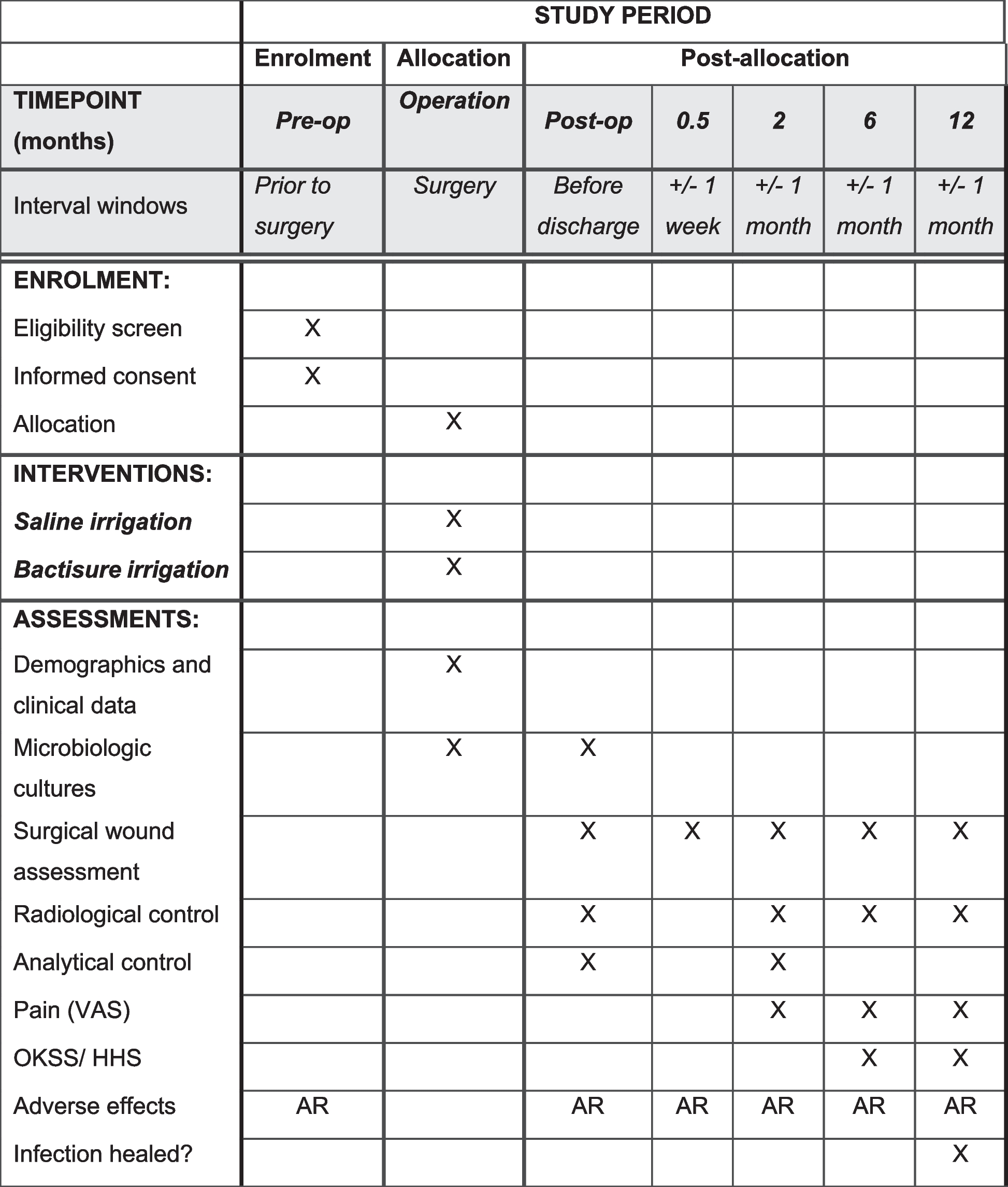
Participant timeline
Enrolment
Post-allocation
* denotes 3-h intervals
Follow-up
Timepoint
Before infusion
1 h
2 h
3 h
6 h
*
12 h
*
24 h
*
36 h
*
48 h
3 months
Enrolment
Eligibility screen
X
Informed consent
X
Randomization
X
Interventions
Dobutamine/placebo (24 h)
X
Assessments
proBNP, biomarkers
X
X
X
X
X
X
BP, P
X
X
X
X
X
X
X
X
X
X
X
X
X
TTE
X
X
RR, SAT, Tp
X
X
X
X
X
X
X
X
X
X
X
X
X
12-lead ECG
X
X
X
X
X
Telemetry
X
Biobank
X
X
X
X
X
X
CMR
X
X
QoL, MoCA, GS, frailty
X
X
Abbreviations:
h hour; proBNP pro-B-type natriuretic peptide, BP blood pressure, P pulse rate, TTE transthoracic echocardiography, RR respiratory rate, SAT saturation, Tp temperature, CMRI cardiac magnetic resonance, QoL quality of life, MoCA Montreal Cognitive Assessment, GS grip strength
*3-h intervals
Sample size
The trial is powered towards the primary endpoint. Recent data on atrial natriuretic peptide (ANP) levels [22], measured 12 h after admission for STEMI patients, were assumed to follow a log-normal distribution with a variation coefficient of 0.59. Assuming an alpha level of 0.05, the trial will achieve a power of 0.86 to show a reduction in proBNP levels of 30% from 1338 ng/L to 937 ng/L if 88 patients is included [23].
ProBNP levels are expected to follow a log-normal distribution with a variation coefficient of 0.59. To account for dropouts and missing data, a total of 100 patients will be enrolled in the trial.
Recruitment
The recruitment period is anticipated to be three years from the enrolment of the first patient. This estimate is based on retrospective analysis of the frequencies of ORBI risk score distribution among STEMI patients and inclusion/exclusion criteria, utilizing data from prior studies conducted at the center [24].



留言 (0)