
記住我
The trial will be conducted at two sites: Steno Diabetes Center North Denmark (Aalborg University Hospital, Denmark) and Steno Diabetes Center Zealand (Zealand University Hospital, Nykøbing Falster).
Eligibility criteriaThe aim is to include 51 participants. The participants will be people with type 2 diabetes on insulin therapy or about to initiate insulin therapy. Participants will be provided with a connected insulin pen from Novo Nordisk A/S, which is compatible exclusively with Novo Nordisk products. Participants using other insulin brands will transition to Novo Nordisk basal insulin (Tresiba®) at trial initiation, following current guidelines for switching insulin. This transition will be managed by highly trained HCPs to ensure safety. A 24/7 hotline staffed by trained personnel will be available to address any issues or questions related to the transition.
Participants will be included according to the following criteria:
Adults ≥ 18 years
Diagnosed with type 2 diabetes ≥ 12 months prior to screening
Willing to travel to one of the two trial sites to attend in-person visits
Have an internet connection at home
Have the personal digital ID, MitID, a Danish national online digital authenticator
Willing to use a smartphone and the connected devices used in the trial
Able to understand and read Danish
Participants will be excluded according to the following criteria:
Pregnancy or breastfeeding
Major surgery planned during the trial
Cancer diagnosis within the last 5 years
Participation in other interventional trials
Limited literacy affecting the use of trial devices
Having worn a continuous glucose monitor (CGM) ≤ 6 months prior to the screening
Treated with mixed insulin
Conditions deemed by the sub-investigator or investigator to make the participant unfit for the trial, including insufficient understanding or physical/cognitive ability to participate.
RecruitmentParticipants will be recruited through a multifaceted approach. Initially, participants will be recruited at the endocrinology clinics at Aalborg University Hospital or Zealand University Hospital, Nykøbing Falster when attending visits. A poster with trial information will also be displayed at the endocrinology clinics, and hospital staff will hand out a leaflet to interested patients. During a visit to the endocrinology clinics, patients can consent to be contacted by a trial representative to receive further information. Additionally, a group of patients has volunteered to be contacted for new trials at the endocrinology clinic at Aalborg University Hospital, and they will receive initial phone calls followed by mailed information if interested. Furthermore, recruitment efforts will extend to newspaper ads, advertisements in the Danish diabetes magazine, social media, patient organizations, and relevant regional websites.
Informed consentPatients expressing interest will be invited to an information interview, where they can bring a companion if desired. Detailed in the participant information letter provided beforehand, this interview outlines the trial’s purpose and design. The interview will be conducted at Aalborg University Hospital or Zealand University Hospital, Nykøbing Falster, in a private setting led by the investigator, a sub-investigator, or a delegated project team member possessing requisite expertise related to the trial. Participants are informed of their right to take time for reflection before consenting, and withdrawal of consent is permitted without explanation at any stage. The trial commences only upon the signing of informed consent by both participant and investigator or sub-investigator.
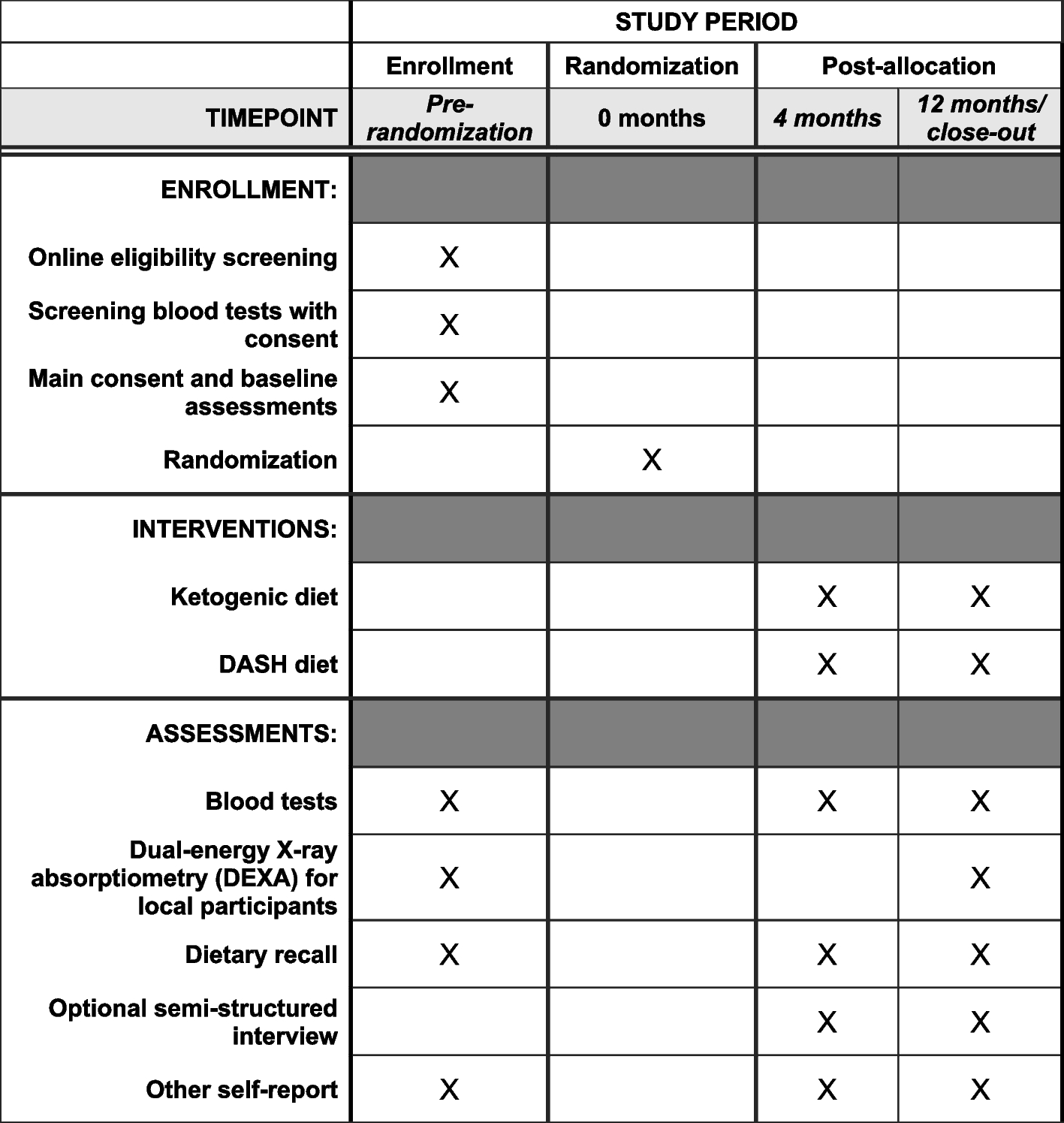
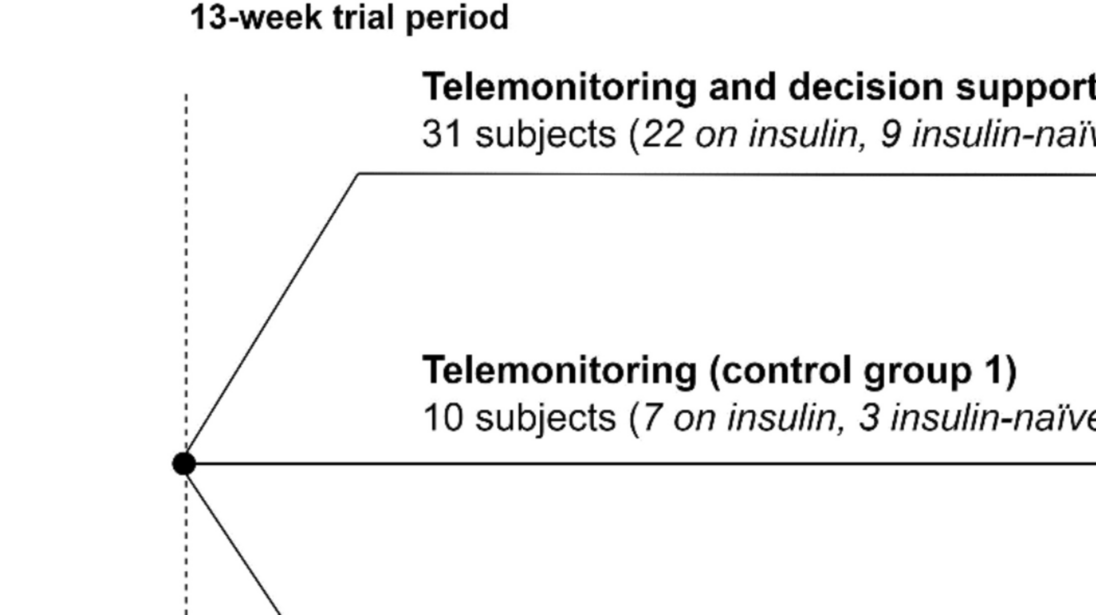
The randomization processIn case of informed consent, the participant will be randomized to either intelligent telemonitoring, telemonitoring alone, or usual care during the initial visit to the endocrinology clinic at Aalborg University Hospital or Zealand University Hospital, Nykøbing Falster. Randomization will be conducted in REDCap (2020 Vanderbilt University) using a computer-generated block-randomized list (3:1:1 ratio) stratified on the proportion of insulin-naïve, resulting in 30% insulin-naïve in each group. The allocations will remain concealed until interventions are assigned. Participants will be enrolled and informed about the trial by HCPs. Participant consent and assignment of interventions will also be handled by HCPs. At the first visit, several basic information about the participants will be obtained together with responses to eight questionnaires (Table 1). Furthermore, a venous blood sample will be drawn to address secondary endpoints and establish baseline data with a lab technician or nurse overseeing the sampling procedure.
Table 1 Overview of assessments, blood samples, and questionnaires collected at trial visits to the endocrinology clinic. *Only participants in the intelligent telemonitoring and telemonitoring group will be asked to answer the DHSS questionnaireThe interventionThe intelligent telemonitoring groupAt the first visit, participants in the intelligent telemonitoring group will be provided with a CGM (Dexcom G7), Novo Nordisk insulin pen(s) (NovoPen6), an activity tracker (Fitbit Charge 4), and a smartphone. Participants treated or initiated with long-acting insulin only will be provided with one insulin pen, while participants treated with long- and short-acting insulin (or will initiate short-acting insulin during the trial) will be provided with two insulin pens. Participants will be asked to use the Novo Nordisk insulin pen(s) to administer insulin for the entire trial period. Participants will also be asked to wear the CGM and an activity tracker for the entire trial period. Participants will use the provided wearable devices continuously at home to collect, log, and transfer tissue glucose levels, insulin administration, activity, and sleep for the entire trial duration. During the trial, participants will be asked to use three smartphone apps: one for activity tracker data (Fitbit: Health and Fitness), one for CGM data (Dexcom G7 app), and one for transferring insulin data recorded by the insulin pen(s) (ADAPT app). Participants will use the provided smartphone to access the apps. Furthermore, participants will be asked to use a web portal (ADAPT portal for patients), which can be accessed via the Internet on a computer. In the web portal, participants will be able to see a combined view of CGM, insulin, and activity data. Participants will therefore need a computer with internet access during the trial to access the combined overview of the data collected from the wearable devices. If participants do not have a computer, they will be provided with one for the trial period. HCPs will train participants in using the technologies at the first visit and provide assistance during the trial.
The telemonitoring system incorporating decision support will support the monitoring HCPs in telemonitoring the participants. An overview of the system and the exchange of data across the parts of the system is seen in Fig. 2. The monitoring HCPs will receive training in using the system prior to trial initiation to equip them to use the system to telemonitor participants.
Fig. 2
Overview of the different parts of the developed decision support system and the exchange of data across these parts from the wearable devices (CGM, activity tracker, and insulin pen) to the patient-facing app on a smartphone to a cloud server form where the healthcare professionals can assess the participants data and update insulin treatment
HCPs affiliated with the clinical trial sites will perform the telemonitoring using the HCP-facing part, ADAPT Portal, of the telemonitoring system which will comprise visualizations of data collected by the wearable devices (CGM, insulin pen(s), and activity tracker) and provide decision support using two algorithms built into the system, a treatment insights algorithm, and a basal insulin dosage suggestion algorithm:
1)The treatment insights algorithm will provide insights into trends in data that may elucidate treatment-relevant issues for a participant’s current treatment (e.g., misalignment between insulin doses taken and the ones prescribed, missed insulin doses, etc.).
2)The basal insulin dosage algorithm will provide the HCPs with a suggestion for basal insulin dose based on the CGM and insulin pen data collected from the participants.
Decision support will also be directed at participants through the ADAPT app, as they can choose to acquire their risk of nocturnal hypoglycemia before going to bed by actively requesting this risk by pushing a button in the ADAPT app. An algorithm will generate the risk. This algorithm is part of the intelligent telemonitoring system.
HCPs, in collaboration with an endocrinologist, make all treatment decisions during the trial. Thus, the intelligent telemonitoring system will only provide information that the monitoring HCPs may choose to act according to if they agree with the treatment suggestion.
The HCPs will initiate telephone contact with participants on at least two occasions—1 week and 1 month after inclusion in the trial. However, the monitoring frequency will be customized to suit each participant’s requirements, and participants may receive ongoing contact if deemed relevant by the monitoring HCPs. All calls to participants are recorded in trial notes, and any treatment-related information from the calls will be recorded in their respective electronic patient journals.
The telemonitoring groupParticipants in the telemonitoring group will be monitored in the same manner as those in the intelligent telemonitoring group. They will also be asked to use a CGM (Dexcom G7), Novo Nordisk insulin pen(s) (NovoPen6), an activity tracker (Fitbit Charge 4), and a smartphone. The only difference between the two groups is that no decision support will be provided to either the participants or the HCPs monitoring participants in the telemonitoring group.
The usual care groupParticipants in the usual care group will receive usual care but will be provided with a blinded CGM (Dexcom G7) with a receiver, insulin pen(s) (NovoPen6), and a smartphone with the ADAPT app. Usual care of people with type 2 diabetes typically covers an annual or biannual consultation. Other contacts with HCPs are on the patient’s initiative and, therefore, the frequency of contacts with HCPs varies greatly from person to person. People with type 2 diabetes treated with or initiating insulin are encouraged to measure blood glucose using a glucometer at least once daily if treated only with basal insulin and at least thrice daily if treated with basal insulin in combination with bolus insulin daily (39). Participants will be asked to wear the blinded CGM during two periods: in continuation of the first visit and before end of trial visit. Each period will have a duration of 20 ± 3 days. These participants will get a receiver to collect the data, as they will not have the Dexcom G7 app. Furthermore, they will be asked to use the insulin pen(s) for the entire trial period and to transfer insulin data to the ADAPT app. Participants will not be able to view their CGM data or insulin data during the trial. No changes in insulin dosage will be performed based on the blinded devices, as neither participants nor monitoring HCPs have access to data. CGM data will be used to analyze primary and secondary outcomes. Recorded insulin data will be used for later analysis and comparison of adherence between the three groups.
Criteria for discontinuingIn case of any serious adverse events related to the trial are identified, as determined by the primary investigator, the trial will be stopped. If a participant reacts unexpectedly to the trial according to the primary investigator’s assessment, or if a participant is otherwise not suited for further participation (e.g., not using the devices as assigned or not attending visits at the endocrinology outpatient clinic), participation can be terminated at any time.
Strategies to improve adherence to interventionIntervention adherence will be assessed using data obtained from the connected devices and from interactions between participants and HCPs.
End of trialAt the end of the trial visit to the endocrinology clinic, participants from all three groups will be asked to answer several questionnaires, and a HCP will perform assessments and take a venous blood sample (see Table 1).
Furthermore, participants who transitioned to Tresiba® from other insulins will be carefully monitored to ensure a smooth transition back to their pre-trial insulin brand if necessary.
Outcome measuresAll CGM-based endpoints are defined using the International Consensus Report from 2019 [25].
Primary endpointChange from baseline in CGM time in range (3.9–10.0 mmol/L) three months after randomization.
Secondary endpoints 1)Change from baseline in HbA1c three months after randomization.
2)Change from baseline in CGM time in level 1 hypoglycemia (3.0–3.8 mmol/L) three months after randomization.
3)Change from baseline in CGM time in level 2 hypoglycemia (< 3.0 mmol/L) three months after randomization.
4)Change from baseline in CGM time in level 1 hyperglycemia (10.1–13.9 mmol/L) three months after randomization.
5)Change from baseline in CGM time in level 2 hyperglycemia (> 13.9 mmol/L) three months after randomization.
6)Change from baseline in total daily dose three months after randomization.
7)Change from baseline in CGM endpoints three months after randomization: episodes (hypoglycemia and hyperglycemia) 15 min and area under the curve.
8)Change from baseline in body weight three months after randomization.
9)Use of telemonitoring equipment: The frequency of use during the trial is assessed directly from the delivered devices.
10)Time-to-target with respect to individualized treatment targets.
11)Time efficiency: Between-group differences in time spent on contact with participants and treatment evaluation/telemonitoring of participants by HCPs during the 3-month trial.
12)Fear of hypoglycemia: Between-group differences measured by the hypoglycemia Fear Survey-II short form (HFS-II short form) questionnaire at baseline and the 3-month assessment
13)Diabetes-related quality of life: Between-group differences measured by the DAWN Impact of Diabetes Profile questionnaire (DIDP) at baseline and the 3-month assessment.
14)Quality of Life: Between-group differences measured by the EQ-5D questionnaire at baseline and the 3-month assessment.
15)Patient adherence: Between-group differences measured by the Morisky Medication Adherence Scale 8-item (MMAS-8) at baseline and 3-month assessment.
16)Participants’ and HCPs’ satisfaction with telemonitoring. Measured by Digital Health Solution Satisfaction questionnaire (DHSS) at the 3-month assessment.
17)Treatment satisfaction: Between-group differences measured by the Diabetes Treatment Satisfaction Questionnaire (DTSQ) at baseline and the 3-month assessment.
18)Perceived competence in Diabetes: Between-group differences measured by the Perceived Competence in Diabetes questionnaire (PCD) at baseline and the 3-month assessment.
Exploratory endpointsThe exploratory endpoints are based on questionnaires and monitoring data and include the following:
19)Self-reported adherence information. During contact between HCPs and participants in the two telemonitoring groups, participants will be asked about their insulin adherence during the previous week.
20)Insulin doses and times. Any differences in the use of insulin between the three groups are examined based on data from the insulin pens.
21)Change from baseline in CGM endpoints three months after randomization: number of days worn, percentage of time active, mean glucose, glycemic variability, and glucose management indicator.
22)Changes from baseline in diet and exercise habits.
23)Self-reported information on diet and exercise habits was collected during contact between HCPs and participants in the two telemonitoring groups.
In addition, qualitative interviews and usability testing will be conducted with selected participants and HCPs from the trial sites to gain deeper insights into the participants’ and HCPs’ experience of the design and use of the intelligent telemonitoring solution and telemonitoring alone.
Data managementData management involves both manual archiving and storage on a computer system within REDCap. The storage duration is in accordance with regulations notified to the Danish Data Protection Agency. Signed informed consent forms are securely stored and kept under lock and key. Questionnaire responses and other patient data are housed within REDCap. Access to all data is restricted, known only to the investigator, sub-investigator, and select project members.
Any review or manipulation of third-party data will occur only with agreement between the sub-investigator and the primary investigator. Data will be retained for the duration specified by the Danish Data Protection Agency. If renewal is requested, the trial will be reevaluated for permission. Data cleaning procedures are carried out throughout the trial, including cross-validation of dates, duplicate searches, and source data verification. Corrections to source data adhere to ICH guidelines for good clinical practice.
ConfidentialityDuring the trial, participant information will be collected, encompassing health status, medication, and comorbidities. This data serves three purposes: ensuring subject suitability for study inclusion, facilitating later analysis in anonymized forms, and aiding HCPs in providing appropriate patient care. Information retrieval occurs only after informed consent is obtained, granting primary investigators, sub-investigators, and designated project members direct access to patient journals, including electronic records. Retrieved data includes health conditions vital for trial completion and for monitoring and quality control purposes. Access to patient information is restricted to HCPs and relevant project team members at Steno Diabetes Centers, with no access granted to personally identifiable data for other ADAPT-T2D consortium members. Regulatory authorities may access patient journals to fulfill monitoring and quality control obligations essential for study completion.
Plans for collection, laboratory evaluation, and storage of biological specimensBlood samples will be drawn from all participants at the initial and the final visit. These samples are analyzed for HbA1c, lipids, c-peptide, glucose, and insulin levels. Approximately 30 mL of blood will be collected per participant. After extraction, the samples are analyzed at Aalborg University Hospital or Zealand University Hospital, Nykøbing Falster and subsequently disposed of. HCPs at Steno Diabetes Center North Denmark and Steno Diabetes Center Zealand will manage the blood collection and processing. All biological material will be discarded once all relevant analyses for the research project are completed at its conclusion.
Sample size calculationThe sample size calculation follows this formula:
$$n=\frac_^}^}_\frac+_\right)}^$$
τ represents the minimum detectable difference in mean, while \(_\) represents the standard deviation of the difference. Assuming a difference in CGM time of 15.9 (τ = 15.9) and a standard deviation of the difference of 15.5% (\(_\) = 15.5) [34], with a significance level of 0.05 and a power of 0.8, the required number of participants is 39. The assumed difference in CGM time in range and the standard deviation of the difference is based on preliminary data from the DiaMonT trial (NCT04981808). Accounting for an estimated dropout rate of 30%, 51 participants need to be included. The high anticipated dropout rate is primarily expected in the usual care group. To mitigate loss to follow-up, HCPs will provide participants in the usual care group with 2 months telemonitoring after trial completion.
Statistical analysisThe trial’s objective will be explored using a range of statistical methods. No formal analysis is planned during the data collection phase.
All analyses will adhere to both intention-to-treat and per-protocol principles. The intention-to-treat analysis will encompass all participants randomized across the trial’s three groups, while the per-protocol analysis will exclude those lost to follow-up.
The primary endpoint will be investigated using data from all participants via a primary analysis utilizing a statistical model incorporating multiple imputation using multivariate imputation by chained equations (MICE). This technique fills in missing CGM data using information from participants in the usual care group (treatment policy). An ANCOVA analysis with CGM time in range at baseline as a covariate will estimate the effect of intelligent telemonitoring on CGM time in range compared to the telemonitoring alone and usual care.
Secondary endpoints, CGM metrics, and HbA1c will undergo similar ANCOVA analyses, also with baseline values serving as covariates. The treatment policy will be employed to impute missing data points.
Sensitivity analyses with similar configurations will be applied to subgroups. Subgroup analyses will differentiate between participants already receiving insulin treatment pre-trial and those who are insulin naïve. Furthermore, the total daily insulin dose will be investigated in a sensitivity analysis among subgroups, and the total daily dose of basal and bolus insulin will be investigated.
Researchers from the project group will conduct the data analysis without blinding to group assignments due to practical and financial considerations.
MonitoringComposition of the coordinating center and trial steering committeeThe ADAPT-T2D consortium’s steering committee will supervise the trial and fulfill the role of the data monitoring committee. Additionally, an independent research member from Steno Diabetes Center North Denmark, uninvolved directly in the trial, will conduct audits on data quality throughout the trial duration.
The core team responsible for managing the trial will include lab technicians and research project managers. They will convene regular meetings as needed throughout the trial period, with monthly status meetings scheduled. The primary investigator, the data auditing research member, and other relevant project members will be invited to these meetings and participate as required.
Adverse event reportingThe risks and potential side effects of participating in the trial are deemed minimal. Any adverse events linked to the trial will be documented in both the electronic patient journal and the trial master file in REDCap if attributed to the trial by the primary investigator or his/her authorized representative. All trial-associated adverse events will be included in the primary trial publication.
留言 (0)