
記住我
This protocol has been written using the SPIRIT reporting guidelines [30].
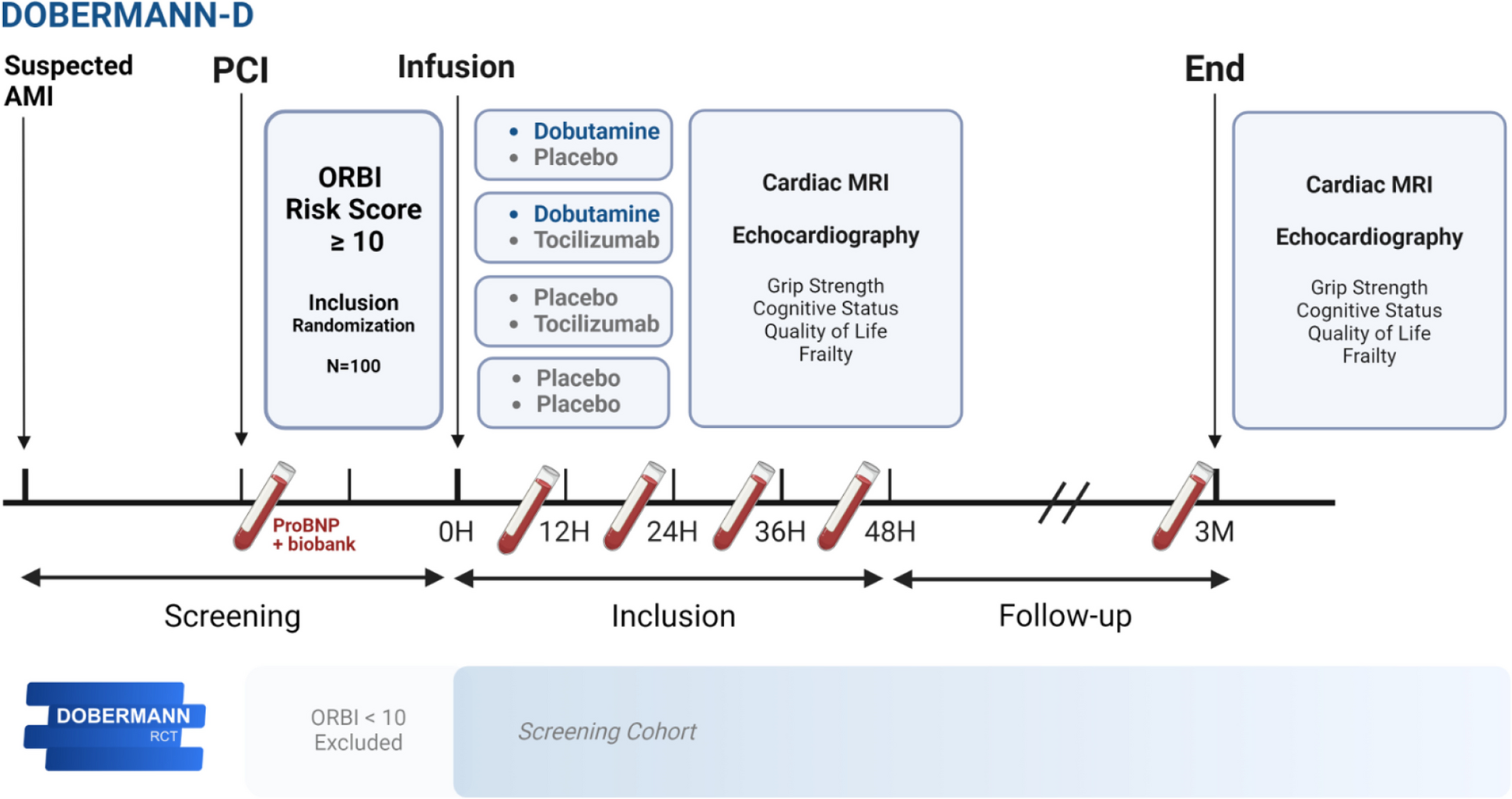
Study designThe study design is a multicentre randomised controlled trial assessing the superiority of paclitaxel-coated or sirolimus-coated balloons compared to an uncoated control balloon. Following a successful plain balloon fistuloplasty, participants will be randomised to further treatment with a paclitaxel-coated balloon, a sirolimus-coated balloon, or an uncoated control balloon. We will recruit 642 patients, each with one or two treatment segments, over a 3-year period. Patients will remain in the trial and be followed up for 1 year.
Treatment segments will be referred to as treatment segments A or B. Treatment segment A is at the anastomosis or is nearer to the anastomosis than treatment segment B. If there is only one treatment segment, this is always treatment segment A.
Primary endpointTime to loss of treatment segment primary patency (TSPP). The treatment segment is the length of the vein that was in contact with a balloon during the index plain balloon fistuloplasty. TSPP is defined as patency with no re-intervention to the area 5 mm proximal to, within, and 5 mm distal to, the treatment segment. TSPP ends when any of the following occurs: (a) clinically driven re-intervention to the treatment segment; (b) thrombotic occlusion considered to be due to restenosis at the treatment segment; (c) surgical intervention that excludes the treatment segment from the access circuit; (d) abandonment of the AVF due to an inability to retreat the treatment segment. In patients with two treatment segments, the primary endpoint will be determined for each treatment segment. The unit of analysis is therefore the treatment segment.
Secondary endpoints 1.Time to loss of primary patency at any treatment segment. In participants with two treatment segments, this is the time to loss of primary patency at either treatment segment.
2.Time to loss of access circuit primary patency. The access circuit is defined as starting at the arterial anastomosis and ending at the cavo-atrial junction. Access circuit primary patency ends when any of the following occurs: (a) access circuit thrombosis, (b) an intervention (either radiological or surgical) anywhere in the access circuit, or (c) the AVF is abandoned due to an inability to treat any lesion.
3.Time to AVF abandonment. AVF abandonment occurs when the AVF is abandoned, regardless of radiological or surgical interventions, with or without a thrombosis event.
4.Total number of radiological or surgical interventions.
5.Adverse events (e.g. thrombosis, infection localised to AVF, rupture of AVF).
6.Patient quality of life as assessed by the mean difference versus the control arm at 6 and 12 months for (a) the EuroQol EQ-5D-5L generic health survey and (b) the vascular access specific VASQoL survey.
7.Mean difference versus control arm at 3 months for (a) intima-media thickness (IMT) and (b) the degree of stenosis measured on ultrasound (only at some sites).
For intima-media thickness (IMT) and the degree of stenosis measured on ultrasound, the unit of analysis is the treatment segment. For the other secondary endpoints, the unit of analysis is the patient.
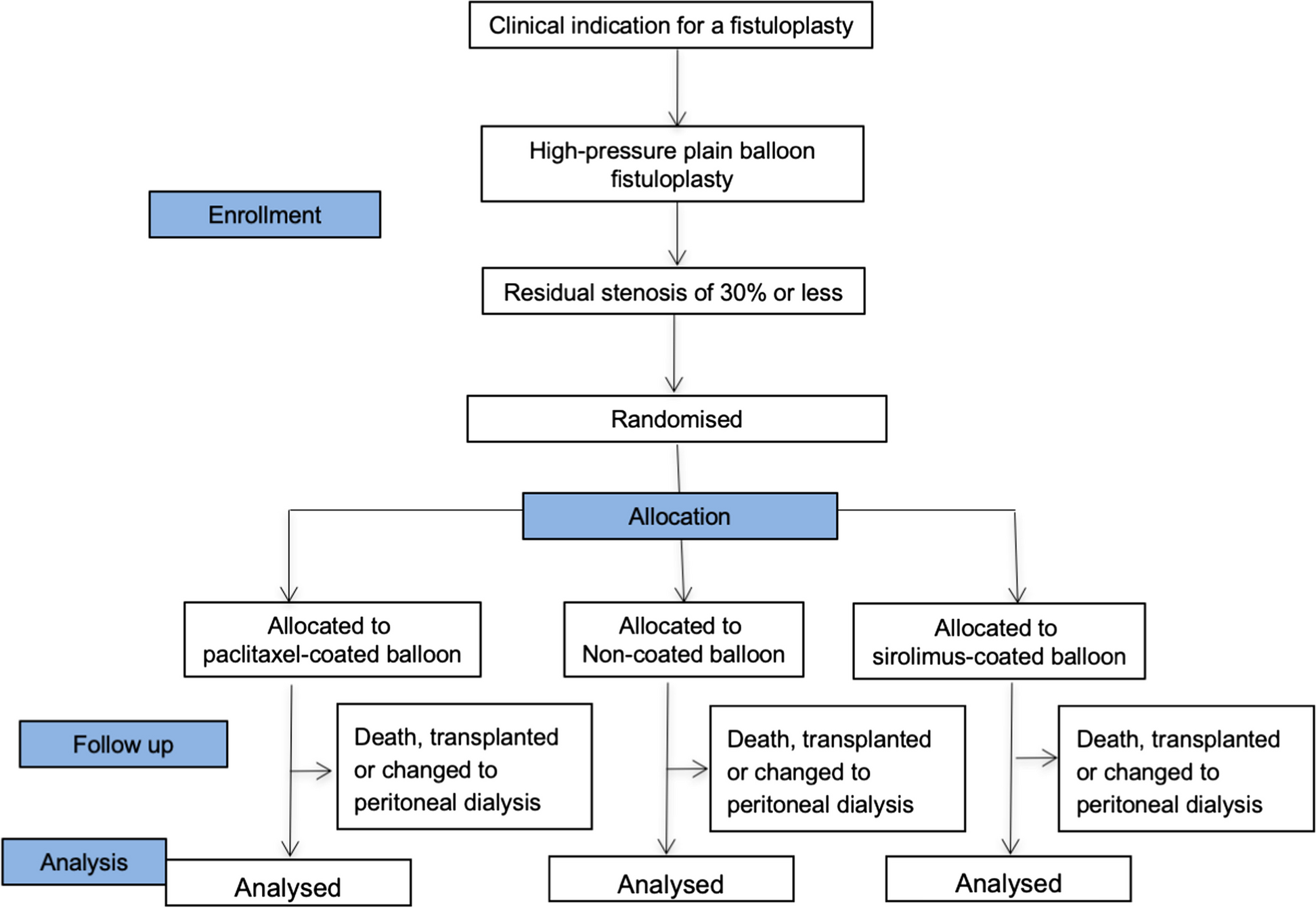
Study flowchartThis is shown in Fig. 1.
Fig. 1 Subject selection
Subject selectionPatients receiving treatment with haemodialysis will be recruited from renal units throughout the UK and at least 20 sites are expected to participate.
Subject inclusion criteria 1.Patients (18 years or over) who have a surgically formed AVF in the arm which has been used for at least 8 dialysis sessions in the preceding 4 weeks.
2.An indication for a fistuloplasty as determined by the local clinical team.
3.The access circuit is free of synthetic graft material or stents.
4.Patient able to give informed consent.
5.Patient willing and able to comply with all study-related procedures.
6.People who are not breastfeeding, not pregnant, not intending to become pregnant or not intending to father children, within 2 years of study treatment
7.No evidence of active systemic or local (to the fistula) infection.
8.No known hypersensitivity or contraindication to contrast medium which cannot be adequately premedicated.
9.No known hypersensitivity or contraindication to paclitaxel or sirolimus.
10.One or two treatment segments. Each treatment segment will contain one or more stenoses of at least 50%.
11.Each treatment segment will be amenable to treatment with a single drug-coated balloon 8 cm in length or two overlapping drug-coated balloons 4 cm in length.
Subject exclusion criteria 1.Thrombosed (failed) access circuit at the time of treatment.
2.Location of a stenosis central to the thoracic inlet.
3.The presence of a lesion that has been treated with a plain balloon fistuloplasty where the diameter of the outflow vein is larger than the size of the largest available drug-coated balloon.
4.The presence of a lesion that has been treated with a plain balloon fistuloplasty where the diameter of the outflow vein is considered too small to be treated with the smallest available drug-coated balloon.
5.A significant residual stenosis (more than 30%) at any treated lesion after plain balloon fistuloplasty.
6.Lack of availability of any of the three types of treatment balloon (Medtronic IN.PACT, Concept Medical MagicTouch or control) at the required size.
Subject recruitmentPatients who may be eligible will be identified by surgeons, specialist nurses, radiologists and nephrologists. Informed consent will be obtained by the delegated clinician(s) at each site, following an explanation of the trial procedures and providing the patient with the Participant Information Sheet. The patient will be given sufficient time to read the information, consider the trial and ask questions. Information will be given at the earliest possible opportunity. Less than 24 h notice should be avoided if possible. Consent will be signed before any study-specific procedures are undertaken. If the patient remains potentially eligible for the study before going to radiology for a fistuloplasty, the treating radiologist will be informed. If possible, the person obtaining consent will not be the radiologist who would administer the study treatment if the patient is randomised. If this is not possible then it will not be considered a protocol violation.
The pre-procedure fistulogramThis will take place immediately prior to the plain balloon fistuloplasty. This will be performed in a dedicated Interventional Radiology suite equipped with digital subtraction angiogram, image overlay/roadmap post-processing capabilities and the ability to capture still and video DICOM file data.
It will be performed through a sheath or cannula placed in the dialysis circuit according to the following specifications: (1) all fistulograms performed as digital subtraction acquisitions at a minimum of 2 frames per second; (2) the entire access circuit from anastomosis to central vein covered in up to 3 stages; (3) medial epicondyle of humerus visible bony landmark on forearm acquisition, acromioclavicular joint on upper arm and central acquisitions; and (4) forearm acquisition to include (i) anteroposterior projection of anastomosis and (ii) oblique projection of anastomosis (specify oblique and craniocaudal angulation).
After the pre-procedure fistulogram, the radiologist will assess all inclusion and exclusion criteria, to decide if the patient remains potentially eligible for the study.
The plain balloon fistuloplasty procedureThis is performed as standard of care. Prior to treatment, 3000–5000 IU of heparin is administered. For all patients, treatment has two components. The fistuloplasty procedure is performed with a high-pressure balloon having a rated burst pressure of > 18 Atm, unless there is a clinical reason to use a different balloon. The following criteria will be met: (1) sized to nominal vein diameter; (2) ensure obliteration of the lesion waist.
Completion fistulogram I is performed after the plain balloon fistuloplasty to ensure adequate therapy according to the following specifications: (1) All fistulograms performed as digital subtraction acquisitions at a minimum of 2 frames per second; (2) Acquisition that demonstrates the treatment segment(s) matched as close as possible to the respective pre-procedure fistulogram acquisition.
After the plain balloon fistuloplasty and completion fistulogram I, the radiologist will review exclusion criteria 5 (a significant residual stenosis (more than 30%) at any treated lesion after plain balloon fistuloplasty) to assess if the participant remains eligible.
Randomisation proceduresRandomisation will be at the level of the individual participants, minimising on (i) study site, (ii) whether the AVF has had a previous radiological intervention, (iii) whether the AVF is in the upper arm or forearm and (iv) whether there are one or two treatment segments.
Minimisation will be implemented using an independent web-based randomisation system hosted at the UKCRC registered clinical trials unit at KCL. Site staff will access the service via www.ctu.co.uk using a computer in the angiography room or an office nearby. It will be performed by the radiologist or their nominee, who will log into the system, enter the participant ID number, initials, date of birth, recruiting radiologist, whether the participant has had a previous radiological intervention in the access circuit and whether the AVF is in the upper arm or forearm. Nominees must not be members of the direct care team making decisions about vascular access, and site research team members who randomise will not be involved in the follow-up of that specific patient. Each randomiser will have unique user access, provided by the CTU upon the authorisation of the trial manager, once the delegation of authority form has been completed. Once randomised, the system will automatically generate a confirmation email to the randomiser and trial manager. In patients with two treatment segments, both will be allocated to the same study treatment.
If it is not possible to use the randomisation system, randomisation may occur using the toss of a coin in order to avoid losing the patient from the study. This should only be needed, if at all, in specific and rare situations such as the CTU server being inaccessible. This will be performed by two people and will require at least two coin tosses as follows. heads then heads = control arm; heads then tails = paclitaxel-coated balloon; tails then heads = sirolimus-coated balloon; tails then tails = toss coin again (twice). This process is repeated as needed until the two coin tosses give an outcome other than tails then tails. The CTU must be informed of the coin randomisation as soon as possible.
Study treatmentIn the intervention arm, the second component is the insertion of a single drug-coated balloon (Medtronic IN.PACT or Concept Medical MagicTouch) or two overlapping 4 cm drug-coated balloons. If two drug-coated balloons are used, they must overlap by at least 5 mm. Drug-coated balloon(s) must be of identical diameter to or 1 mm bigger than the largest diameter high-pressure plain balloon used. The length of the drug-coated balloon or overlapping drug-coated balloons must be a minimum of 1 cm longer (5 mm at either end) than the entire segment of the vein that has been in contact with a high-pressure plain balloon.
The drug-coated balloon(s) will be inflated to nominal pressure for a minimum of 180 s duration. The duration of inflation will be documented in the eCRF. Instructions for the use of the drug-coated balloon are stringently adhered to ensure appropriate preparation and handling of the device.
In the control arm, an identical procedure is followed, but using a control balloon that is not drug coated. The diameter and length requirements of the control balloon are also identical. Wherever possible, the Medtronic Admiral Xtreme balloon will be used. However, if this is not available, a similar control balloon may be used, and this will not be considered a protocol violation.
In both arms, image overlay/roadmap will be utilised to ensure that there is no geographical mismatch between the segments treated with plain balloon or the study treatment balloon. A completion fistulogram is performed (completion fistulogram II) to confirm no angiographically visible effect after treatment with the drug-coated or control balloon, according to the same specifications as completion fistulogram I.
Measures to avoid biasA fully blinded trial is not possible due to the differing appearances of the balloons. People who will be aware of the treatment allocation include the treating radiologist and the trial managers (for monthly balloon re-stocking purposes). The patient, other radiologists, the direct care team making decisions related to vascular access, the site research team following up with the patient and the trial statistician undertaking the primary efficacy analysis will remain blinded to treatment allocation. If possible, the person obtaining consent will not be the radiologist who would administer the study treatment if the patient were randomised, as described under subject recruitment. Referral for a repeat procedure will originate from the direct care team who will be unaware of treatment allocation.
A different radiologist to the one performing the index procedure will be involved in subsequent clinical decisions and/or perform a repeat procedure when possible. However, it is not possible to guarantee this. Therefore, the radiologist involved in subsequent decisions and/or performing a repeat procedure may have knowledge of whether the patient was treated with a particular drug-coated or uncoated balloon.
Primary endpoint adjudication: The following applies to any patient while they have at least one treatment segment with primary patency maintained (i.e. a treatment segment that has not yet met the primary endpoint of the trial). (a) For any radiological intervention, the data file(s), in addition to the data file(s) from the index procedure, will be sent to the lead study site with the patient’s name replaced by the trial ID. Files will include the pre-procedure fistulogram, fistuloplasty and all completion fistulograms. The images will be reviewed by a radiologist in the research team who is not based at the same site as the patient. They will decide if they agree with the data on the eCRF (electronic case report form) regarding treatment segment primary patency. Any discrepancies will be discussed with the local radiologist in order to reach agreement. (b) The review process described in (a) will also be followed for any fistulogram performed without a subsequent intervention (see fistulograms performed for a clinical indication). (c) For any surgical intervention or abandonment of the AVF, the eCRF will be reviewed by a member of the research team who is not based at the same site as the patient. They will decide if they agree with the data on the eCRF regarding treatment segment primary patency. Any discrepancies will be discussed with a local investigator in order to reach agreement.
Based on the interim analyses, the DMC and TSC may recommend unblinding the data. The statistician undertaking the primary efficacy analysis will remain blinded.
Follow up proceduresStudy visits will occur every 3 months ± 1 month. These visits will take place either face-to-face or via a telephone conversation. Any face-to-face meetings will usually coincide with dialysis to avoid additional patient travel. At study visits, participants will be asked about any access circuit interventions and changes to peritoneal dialysis, renal transplantation or adverse events. Clinical records will also be reviewed.
EQ-5D-5L and VASQoL will be completed at baseline, 6 months and 12 months. These may be administered in person or over the telephone. If the fistula treated at randomisation is no longer in use, participants will be asked to consider the questions in VASQoL in relation to whatever form of access is in use at the time. Patients may decide they do not wish to be contacted for further study visits, but this does not require withdrawal from the study. They can remain under follow-up with relevant data collected from their medical records. Patients will remain in the trial and be followed up for 1 year.
During follow-up, participants may receive a transplant, change from haemodialysis to peritoneal dialysis, or have their AVF abandoned. They will be considered censored for any AVF patency outcomes that have not been reached at that point but will continue under follow-up in order to collect other data. An exception is a transplant that does not function which results in the patient continuing haemodialysis. In this case, censoring for AVF patency outcomes will not occur.
Ultrasound assessmentsPatients at selected sites will be asked to undergo ultrasound assessments. If they decline, then this will not be considered a protocol violation. Three scans will be performed for each treatment segment. These will be (i) 30 days or less, prior to the study intervention, (ii) immediately post intervention and (iii) at 3 months (± 1 month). A high-resolution linear array transducer will be used. B-mode ultrasound images will be acquired at the points of stenosis or at the site of the previous balloon angioplasty, when restenosis has not been demonstrated and will be used to quantify IMT. Measurements will include the outer-to-outer wall vessel diameter and luminal diameter. Volume flow will be measured in the brachial artery and peak systolic velocity will be measured at points of stenosis within each treatment segment. A standard operating procedure will be available and will be followed.
Fistulograms performed for a clinical indicationIn patients with at least one treatment segment that has not yet reached the primary endpoint of the trial, the fistulogram (pre-procedure if there is a subsequent intervention) will follow the same specifications as the pre-procedure fistulogram. Data file(s) will be sent to the lead site for review as specified for primary endpoint adjudication (see measures to avoid bias), even if an intervention is not performed.
End of study definitionEnd of study is defined as the last participant’s last follow-up. The trial may be prematurely discontinued by the Sponsor, Funder, Chief Investigator or TSC based on new safety information or for other reasons given by the DMC, TSC, and REC. The trial may also be prematurely discontinued due to lack of recruitment or upon advice from the TSC who will advise on whether to continue or discontinue the study and make a recommendation to the sponsor. If the trial is prematurely discontinued, active participants will be informed, and no further participant data will be collected.
Blood testsNo blood tests are required as part of the trial. The most recent pre-procedure full blood count and CRP results will be entered on the eCRF.
Assessment of safetyThe PAVE-2 protocol does not fall within the Clinical Trial Regulations and therefore is not a drug trial. In addition, the drug-coated balloons are CE-marked medical devices, so prior regulatory approval from the MHRA is not needed. Safety reporting will be in keeping with the requirements for research other than Clinical Trials of Investigational Medicinal Products.
A Serious Adverse Event (SAE) is an untoward occurrence that (a) results in death, (b) is life-threatening, (c) requires non-elective hospitalisation or prolongation of existing hospitalisation, (d) results in persistent or significant disability or incapacity, (e) consists of a congenital anomaly or birth defect or (f) is otherwise considered medically significant by the investigator.
For this trial, SAEs will only be reported if they are related to the study treatment or the access circuit that has been treated and if they are unexpected. Examples of events that will not be considered SAEs include (but are not limited to) the following: Respiratory infections, fluid overload, diverticulitis, gastrointestinal infection, bowel obstruction, cholecystitis, falls, syncope, myocardial infarction, fractures, foot infections, skin infection (unrelated to the access circuit), urinary tract infection,
We do not expect any SAEs to be related to the study treatment or the access circuit that has been treated. Therefore, any SAEs that are considered to be related to the study treatment or the access circuit that has been treated will be reported. They will be reported by the local investigators on the SAE form to the Chief Investigator, as soon as they become aware and within 24 h at most. Although it is not an SAE, any pregnancy or fathering of children that occurs during follow-up will be reported via the SAE system.
Since the study treatment is local and not systemic, non-serious adverse events will be defined as events that local principal investigator (PI) considers are directly related to the study treatment or the access circuit that has been treated. These should be recorded throughout the trial and will be captured in the eCRF at each study assessment. Deaths of study participants will be recorded. All collected SAEs and AEs will be reported in the primary trial publication.
Ethics reportingReports of SAEs will be reviewed by the CI within 24 h to see if the local PI considers that the event is related to the research procedure and unexpected (a SUSAR) and if so, it will be onward reported to the REC and DMC within 15 days.
Data monitoring committeeThe membership will be decided by the CI and approved by the NIHR. The DMC includes a statistician and two other independent experts. They will receive a report of recruitment, serious and non-serious adverse events and a summary of accumulated clinical data from the trial statistician, and will meet in person, online, or by telephone. They will report to the TSC and will meet at least annually during the study. Additional meetings may take place at the time of interim analysis or in case of recruitment issues. The DMC is advisory to the TSC. The DMC charter will be drafted and agreed prior to recruitment. A Trial Statistician will prepare reports for the DMC.
Trial steering committeeThe TSC membership will be decided by the CI and approved by the NIHR. The chair will be an independent expert. Members will include the CI and a patient representative. At least 75% of members will be independent. The TSC will meet at least annually during the study. Additional meetings may take place at the time of interim analysis or in case of recruitment issues. The TSC is an executive committee. Terms of reference of the TSC will be agreed upon and documented prior to the start of recruitment. The Trial Manager will prepare reports to the TSC.
Ethics and regulatory approvalsThis protocol and related documents were reviewed by the London-Hampstead Research Ethics Committee (REC). HRA and Health and Care Research Wales (HCRW) approval was given (23/LO/0625).
Subject complianceSubject compliance is not expected to be an issue as the study treatment is administered at one time after randomisation.
Subject withdrawalParticipants have the right to withdraw from the study at any time for any reason. It is understood by all concerned that an excessive rate of withdrawals can render the study uninterpretable; therefore, unnecessary withdrawal of patients should be avoided. Should a patient decide to withdraw from the study, all efforts will be made to report the reason for withdrawal as thoroughly as possible. Patients may remain in the trial with no further contact from the research team (see above under follow up procedures). Therefore, we anticipate that withdrawal should be very uncommon.
Protocol complianceAny instances where the allocated treatment is not administered will be promptly reported and investigated to establish the reason and minimise future occurrences. Following randomisation, there are no restrictions on usual care.
Data to be collectedData to be collected at each visit is indicated in the schedule of treatments (Table 1). Sources will include the clinical notes and discussion with the patient. At baseline, the following definitions will be used in the medical history; coronary artery disease is defined as previous myocardial infarction, coronary artery bypass surgery, percutaneous coronary artery intervention or significant disease on a coronary angiogram. Peripheral vascular disease is defined as previous surgery (bypass/amputation), radiological intervention, or evidence of disease on angiography/ultrasound. A stroke will be assumed to have been ischaemic if it is uncertain whether it was ischaemic or haemorrhagic.
Table 1 Schedule of treatments for each visitData handling and record keepingDuring the study, only the direct care team or site research team will have access to participants’ identifiable data. Any paper documents with personal data will be held in a locked filing cabinet in a locked office and retained for a minimum of 5 years following the end of the study.
Pseudonymised clinical and research data for the study will be stored on the eCRF system, hosted at the King’s Clinical Trials Unit, KCL for at least 15 years. The eCRF (InferMed MACRO) is GCP and FDA 21 CFR Part 11 compliant. Data entry staff at site will be provided with unique usernames and passwords to the system and will be trained in data entry by the trial manager. The trial manager will visit sites to review data on the system, raise discrepancies and confirm source data verification checks. All requests for access to the data entry system must be authorised by the trial manager. All requests for data exports must be authorised by the trial statistician. The trial manager will work with the CI and the trial statistician to ensure data is checked and cleaned on an ongoing basis and will confirm all data checks have been completed before database lock.
The investigators and the institutions will permit trial-related monitoring, audits, REC review, and regulatory inspections (where appropriate) by providing direct access to source data and other relevant documents (i.e. patients’ case sheets, blood test reports, and X-ray reports). Record keeping will be the responsibility of the investigators.
留言 (0)