
記住我


This experimental analysis investigates the role of mechanical power (MP) in generating VILI in large, healthy animals. Given the absence of pre-existing lung disease (healthy baseline), the progression observed over time can be attributed to mechanical ventilation, implying the development of VILI. This study has identified a specific power threshold that results in histologically confirmed lung injury.
The main results show that, with an applied MP ranging from 2.6 to 63.4 J min−1 (0.1–2.3 J min−1 kg-1), injury occurred when the applied MPratio exceeded approximately 4.5 times its normal expected value. This normal MP value was computed using data from De Robertis et al. [14] for domestic pigs, similar in weight to our population. The median value of normal expected MP was 0.094 J min−1 kg−1, close to the value expected in normal human (i.e., 6–7 J min−1 in a 70 kg individual) [15].
The two groups of animals defined as having a lower or higher MPratio than threshold of 4.5 differed considerably in terms of respiratory and hemodynamic physiology during the experiment. These differences were also associated with the higher fluid balance needed to maintain the hemodynamics in the group at higher MPratio [16]. Over time, from 0.5 to 48 h, key marker variables diverged between the two groups (Table E4) with the high MPratio group showing increased physiological dead space and worsened respiratory mechanics. These data are consistent with the appearance and worsening of structural lung lesions in that cohort. Despite increased histologically identified atelectasis, venous admixture decreased in the high MPratio group. This is not surprising, considering that atelectasis due to VILI is easily recruitable during inspiratory phase [17] and that oxygenation variables are not closely related to VILI-related outcome, as shown in the ARMA trial [6].
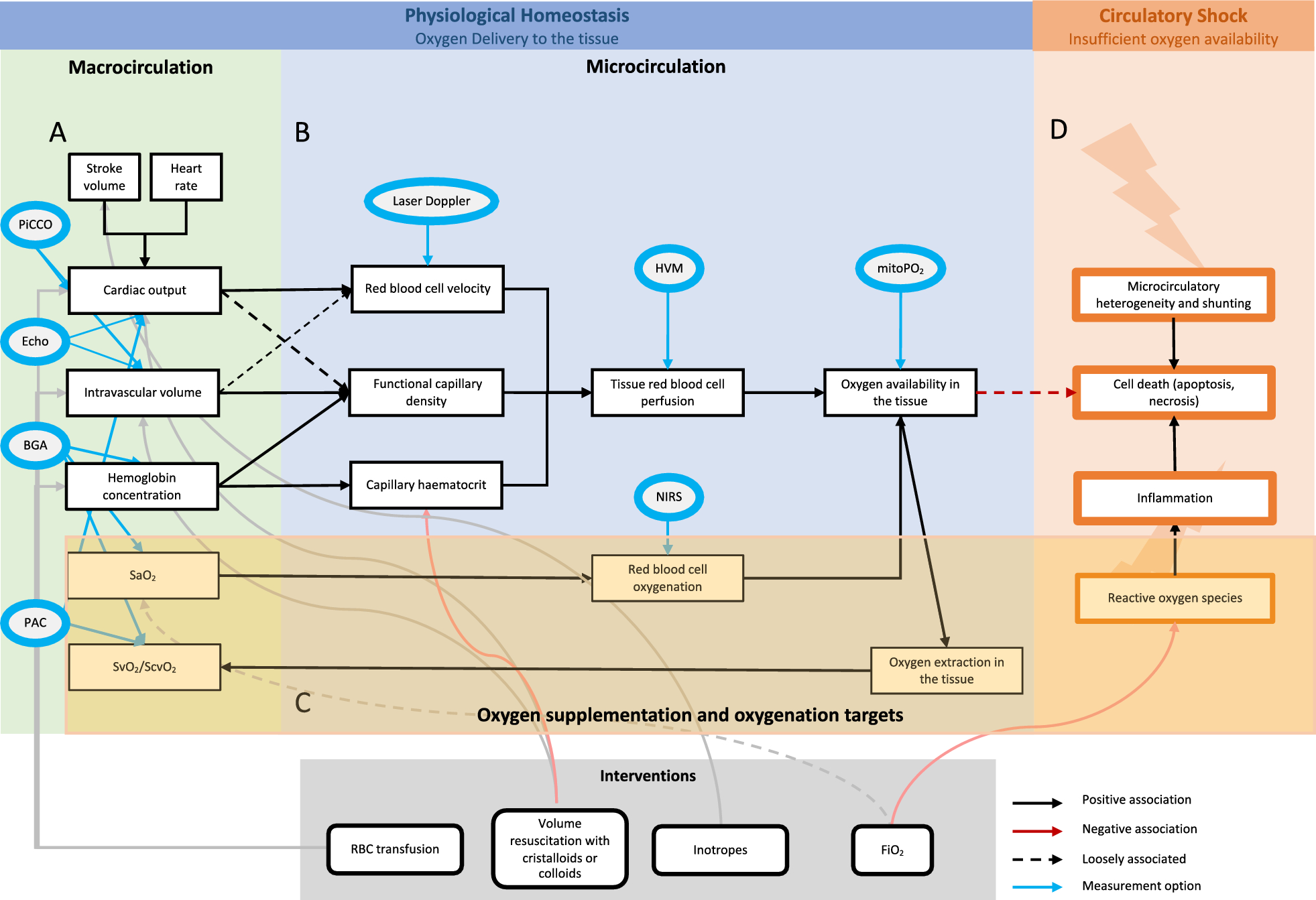
This study highlights the ambiguity in defining VILI, as it remains unclear which variables (anatomic, hemodynamics, mechanics, and gas exchange) are direct expressions of VILI or merely associated damage co-factors. Indeed, some authors have included the hemodynamic alterations and fluid balance under the VILI umbrella, while some others have not [18, 19]. Indeed, the use of terms as ventilator-induced lung injury or ventilation-induced lung injury or ventilator-associated lung injury reflect the lack of clarity of this concept. A possible model for VILI development during mechanical ventilation in healthy lungs [20] may consist of two sequences (Fig. 5).
Fig. 5
Possible sequences leading to ventilator-induced lung injury in healthy lung. Sequence 1, the intensity of mechanical ventilation is such as to induce structural anatomical modifications, which in turn lead to inflammatory reaction. The physiological variables in theory more associated with these alterations should be the physiological dead space and the inflammatory cytokines, both not specific for lung injury. Sequence 2: the intensity of mechanical ventilation is not such as to induce anatomical damage, but sufficient to interfere with normal hemodynamics. In theory, the first event is the increased intrathoracic pressure followed by all the hemodynamic consequences. The estimate of change of intrathoracic pressure requires the esophageal balloon, while several methods are available to assess the hemodynamic status. Both “direct” and “indirect” lung injury sequences convey in the same final pattern, characterized by water retention, alveolar edema and atelectasis. In the figure we report the common physiological variables which should be more associate with these events
Sequence 1: structural alterationsThe gold standard for VILI should be the alveolar ruptures and extracellular matrix fragmentation due to the excessive MP, leading to increased dead space and lung elastance [20], as demonstrated in this study in the high MPratio group. Inflammation: alveolar ruptures and extracellular matrix fragmentation led to marked inflammation, with increased capillary permeability. This phenomenon may be tracked by changes in the level of inflammatory cytokines [21], and by a marked increase of inflammation as observed in histological samples of high MPratio group. Water retention/pulmonary hypertension/pulmonary edema/increased lung weight: Several variables, are associated with these phenomena. Water retention may be estimated by fluid balance, which was clearly more positive in the high MPratio group than in the low one. Lung weight (suggesting lung edema) was also markedly greater in the high MPratio group and was significantly associated with the fluid balance and with structural histological changes of the lung. It appears, therefore, that the formation of lung edema depends on both functional and structural changes of lung parenchyma. Compression atelectasis: lung edema and increased lung weight inevitably lead to compression atelectasis [22, 23]. The best way to measure it is the assessment of recruitability, as only atelectatic (i.e., compressed but empty) pulmonary units may be recruited. Unfortunately, we did not measure recruitability in our experiments. However, the frequency of atelectasis detected by histology was significantly higher in the high MPratio group. The consequent atelectrauma possibly contributed to enhance the inflammatory reaction and its consequences.
Sequence 2It is possible that “harmful mechanical ventilation”, although not so severe as to induce structural changes, is sufficient to induce a marked hemodynamic response. Indeed, in our study we found that mechanical ventilation may be associated with increased pulmonary artery pressure. To support hemodynamics in the high MPratio group, fluid balance was markedly higher, with consequences similar to what is described in sequence 1. In essence, the difference between the two sequences is the lack of structural alteration, which selectively characterizes sequence 1. In our study, we had clear signs of hemodynamic compromise which appear more than a threshold-linked phenomenon, as they appeared proportional to the MP.
LimitationsUsing healthy animals to study ventilator-induced lung injury (VILI) might be seen as a limitation. However, in our research, we consistently use healthy subjects to ensure that the only variable affecting lung damage is the mechanical ventilation per se, free from the confounding factors found in the “ARDS models”. In addition, over the course of our experimental model factors like inhomogeneity and stress raisers develop progressively, aligning the model more closely with the temporal evolution of the ARDS pathophysiology.
This study presents several other limitations primarily due to the retrospective design, which includes data from several experiments conducted at different times. The data derived from three distinct studies, each with potentially different use of fluid and vasopressors. This heterogeneity might have affected the results of the cluster analysis, potentially categorizing animals into groups based on the conditions of one larger study that employed significantly higher mechanical power and fluid balance. Additionally, the distribution of mechanical power ratio (MPratio) may have been more discrete than continuous due to the nature of the experimental design.
Another possible limitation may be the lack of comparisons of MP ratio to other possible “VILI markers” as driving pressure and the index proposed by Costa (4DP + RR) in which DP is the driving pressure and RR is the respiratory rate [24]. However, both driving pressure and frequency are included in the mechanical power equation and therefore these express the same elements of VILI. The direct comparison between mechanical power and driving pressure or 4DP + RR may be the focus of a different study but this may be inappropriate in an experimental model as the specific weighting of DP and RR may be different from the 4:1 found in human clinical trials.
However, we think that despite these limitations the results are valid. Variations in the expected MP do not compromise the validity of identifying a VILI threshold. The distribution of MP and MPratio seems to be near-continuous and well distributed across the range of MPratio (see Figure E4), and the fluid overload was the consequence of the high MP, rather than the cause.
The challenges of assessing VILIThese results and limitations reflect a longstanding debate in the understanding of VILI, which was already raised in the early descriptions of ARDS. Should the physiological hemodynamic response and its consequences (e.g., positive fluid balance and water retention due to the kidney response) be considered VILI in the absence of structural alterations?
Healthy lungWe argue that if one starts with healthy animals, whatever alterations we found were likely due to mechanical ventilation itself, as no prior inflammation, pulmonary hypertension or edema were present prior to its initiation. Accordingly, water retention, fluid balance and hemodynamic compromise should be considered part of VILI. Perhaps, this condition is better described by a term such as ventilator-induced injury, as most vital extrapulmonary organ systems, are indirectly compromised by mechanical ventilation.
Diseased lungWhile the bulk of our results strongly suggest that a distinct MPratio greater than 4.5 causes VILI to become evident, this threshold cannot be, as such, translated to diseased lung and to human ARDS. Indeed, in diseased lung the damage related to the energy input should ideally be normalized to the size of the ventilatable lung, i.e., to the size of baby lung. While inflating the respiratory system requires a level of energy related to its mechanical characteristics, the capacity of the baby lung to receive it is less. The specific energy delivered per lung unit is an inverse function of baby lung size.
留言 (0)