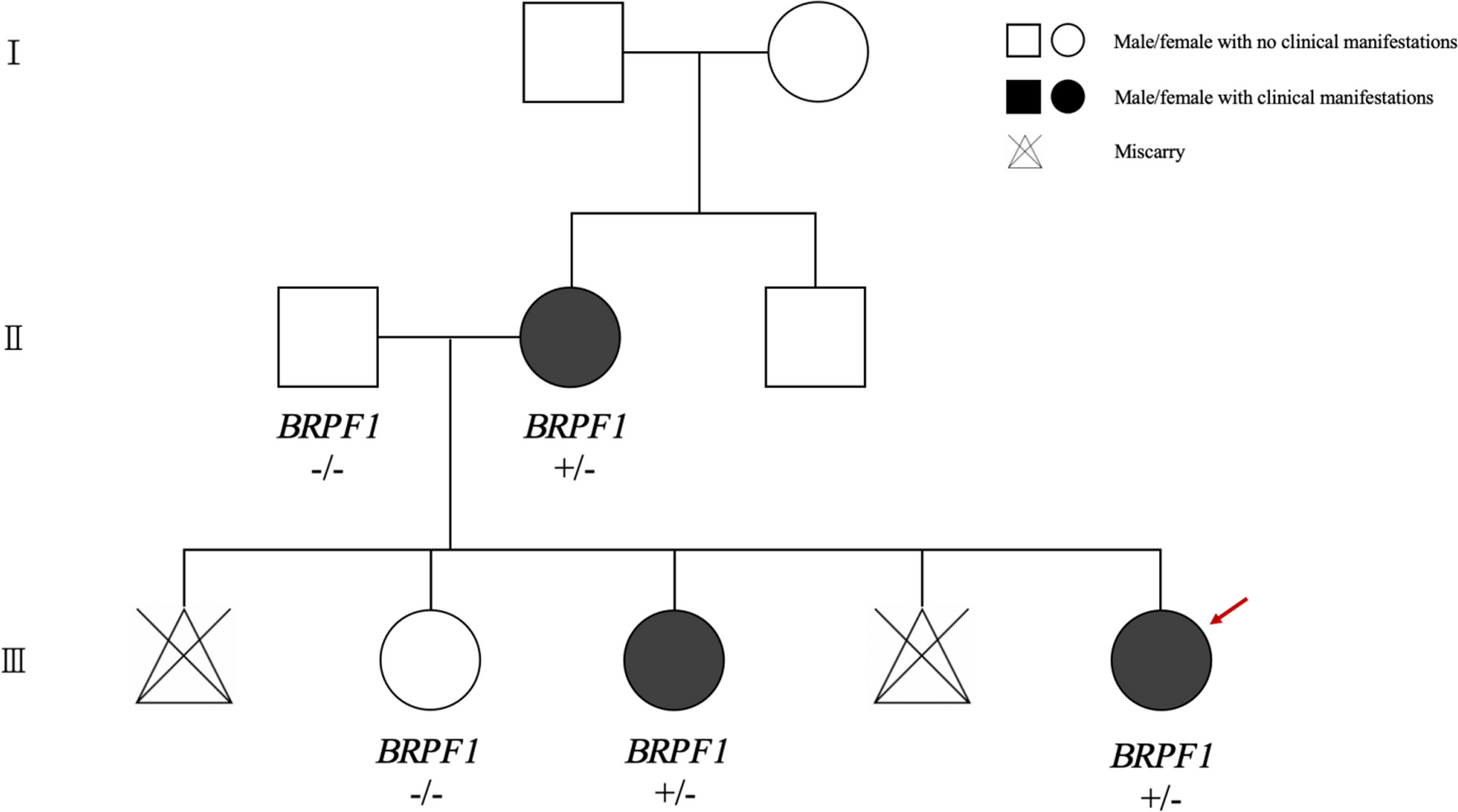
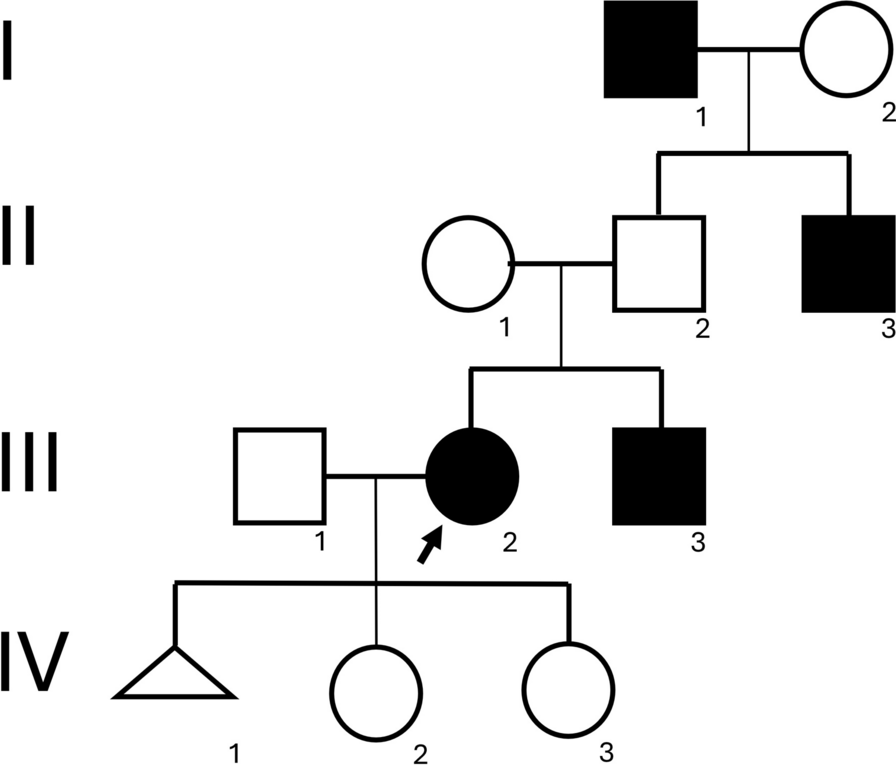
In this study, we report a case involving a Chinese family exhibiting mild ID and facial dysmorphism. The proband, a 10-month-old child, not only displayed facial dysmorphisms but also experienced focal seizures and developmental delays. Genetic testing identified heterozygous deletion variants of BRPF1 in the proband, her mother, and her sister.
BRPF1 variations are associated with IDDDFP, with 46 patients documented across 10 studies (Table 1). The affected individuals typically present with varying degrees of ID (98%, 44/45) and facial deformities, mainly ptosis and blepharophimosis (72%, 32/44). Visual or ocular issues (64%, 25/39), such as strabismus, amblyopia, or refractive error, are relatively common. Additionally, some patients exhibit skeletal deformities (72%, 31/43), including hand (brachydactyly and brachymetacarpia) and foot (clubfoot or syndactyly) differences. However, a minority of patients with IDDDFP experience seizures (21%, 9/43) and structural brain abnormalities (50%, 10/20), often characterized by abnormal white matter signals under MRI and corpus callosum thinning [3, 5,6,7,8,9,10,11,12,13]. The affected family members in our report exhibited typical facial dysmorphisms of IDDDFP. Specifically, the proband, mother, and sister exhibited facial features consistent with hypoblepharoptosis. Owing to the proband’s young age, an intelligence evaluation was not conducted. However, both her mother and sister exhibited mild ID without signs of developmental delay. While epilepsy is observed in a minority of patients with BRPF1 defects, onset typically occurs in childhood [5, 6, 10]. Notably, only one case involving a 3-month-old infant with fever-induced multiple seizures has been documented, similar to the seizure characteristics in our proband, suggesting the involvement of BRPF1 in the mechanism of epilepsy [10].
Table 1 The phenotypes and genotypes of the three patients in this study are related to those reported in patients with IDDDFPThe primary reported variants comprised frameshift (47%, 16/34), nonsense (26%, 9/34), and three deletion (9%) variants, suggesting haploinsufficiency of BRPF1 as a pathological mechanism [3, 5,6,7,8,9,10,11,12,13]. However, phenotypic variations exist among affected individuals within the same family. For instance, in the family reported by Pode-Shakked et al., all affected offspring exhibited ID despite their mothers carrying the same variants without showing cognitive impairments [3]. Similarly, in the family reported by Mattioli et al., the patients’ offspring exhibited agenesis of the corpus callosum and attention deficit hyperactivity disorder; however, their mother lacked such symptoms [6]. Including the family with ID reported in this study, only the proband exhibited seizures and developmental delay. This emphasizes that BRPF1 defects have variable phenotypic characteristics in neurodevelopmental disorders, suggesting no definitive correlation between the BRPF1 genotype and phenotype. For clinical diagnosis, consideration should be given to IDDDFP in patients with facial deformities, particularly ptosis and blepharophimosis, alongside ID. However, the differential diagnosis should also include Arboleda–Tham syndrome (OMIM#616268) or Say–Barber–Biesecker–Young–Simpson syndrome (OMIM#603736), stemming from KAT6A or KAT6B gene defects, respectively. Patients with KAT6A or KAT6B deficiency may exhibit more severe neurodevelopmental disorders and multisystem malformations [18, 19]. It is also worth noting that CDK13 gene defects lead to congenital heart defects, dysmorphic facial features, and intellectual developmental disorder (OMIM#617360), which is accompanied by facial features such as a wide eye distance, epicanthus, ptosis, and ear deformities; different degrees of nervous system abnormalities; and characteristics of congenital heart disease with high penetrance [20].
BRPF1, located in the p25.3 region of chromosome 3, encodes the BRPF1 protein, comprising 1,214 amino acids. This protein exhibits ubiquitous expression, particularly in the testis and spermatogonia, where it attains the highest expression level [21]. Structurally, the BRPF1 protein comprises three main domains: a bromodomain, a plant homeodomain finger, and a chromo/tudor-related Pro-Trp-Trp-Pro (PWWP) motif [22]. Serving as a crucial component of the KAT6A (also known as MOZ or MYST3)/KAT6B (also known as MORF or MYST4) histone acetyltransferase (HAT), the BRPF1 protein functions as both a scaffolding subunit and transcriptional regulator [23]. Notably, KAT6A and KAT6B defects can result in neurodevelopmental disorder phenotypes similar to those observed in IDDDFP [24, 25]. Previous studies have shown that BRPF1 primarily controls H3K23 acetylation through KAT6A/KAT6B, thereby influencing epigenetic regulation and developmental programs [26, 27]. In a study by Yan et al., BRPF1-deficient patients with neurodevelopmental disorders exhibited disrupted propionylation processes in addition to H3K23 acetylation regulation [10]. This finding suggests that BRPF1 deficiency may regulate the H3K23 acylation process through KAT6, contributing to the pathogenesis of neurodevelopmental disorders. The heterozygous deletion of exons 2–14 in BRPF1 identified in our study impairs the biological functions of the various domains of the BRPF1 protein.
Currently, no effective treatment is available for IDDDFP. However, research by Yan et al. suggested that BRPF1 variation contributes to H3K23 acylation defects. They revealed that valproate, vorinostat, propionate, and butyrate can enhance H3K23 acylation at the cellular level. Specifically, propionate has been shown to enhance H3K23 propionylation in embryonic fibroblasts in mouse models [10]. Understanding these mechanisms and further pharmacological studies may provide potential treatment strategies for patients with IDDDFP. In addition, patients with IDDDFP may be subjected to potential social discrimination due to their ID and appearance, and their quality of life is often low, necessitating special education and psychological counseling.

留言 (0)