Pheochromocytomas are rare tumors of chromaffin cells derived from the neural crest and mainly found in the adrenal medulla, although they can appear in other sites (paragangliomas). These are difficult to diagnose catecholamine-producing neuroendocrine tumors, and the familial aggregation of cases is quite uncommon. Most pheochromocytomas are isolated and apparently sporadic, while approximately one in ten cases occurs in the context of a syndrome (neurofibromatosis type I, Multiple Endocrine Neoplasia type II, Von Hippel–Lindau (VHL) syndrome), sometimes displaying Mendelian patterns of inheritance [1, 2].
The annual incidence of pheochromocytomas is estimated around 2–8 cases per million [3, 4], being responsible for less than 0.1–0.2% of hypertension cases without sex preference, and most are diagnosed between the fourth and fifth decade of life, although they can appear at any age [4, 5].
About 80–85% of pheochromocytomas are found in the adrenal medulla, and the other 15–20% in extra-adrenal areas, mainly the Zuckerkandl organ, although it can occur in other areas of the thorax and abdomen; extra-adrenal pheochromocytomas are more common in children [6, 7].
Pheochromocytomas are classified as benign or malignant, the difference being that the malignant type is capable of metastasizing [1, 3, 5]. The formation of pheochromocytomas has been linked to germline mutations in more than 20 loci, causing activation of three signaling pathways: (1) kinase signaling-related genes, (2) pseudohypoxic Krebs cycle-related genes and (3) Wnt signaling-related genes [1, 6].
The most commonly occurring symptoms in pheochromocytoma cases are blurred vision, chest and abdominal pain, constipation, diaphoresis, fatigue, fever, flushing, headache, heat intolerance, hyperglycemia, hypertension, nausea, palpitations, pallor, panic attacks and anxiety disorders, papilledema, polyuria and polydipsia, sweating, vomiting, tremors and weight loss [2, 3, 5, 6].
Diagnosis is based on confirmation of clinical suspicion by biochemical tests and image studies [2, 8]. Biochemical tests are indicated in asymptomatic patients with family history of pheochromocytoma or germline mutations associated with increased risk for pheochromocytoma as well as patients with either (1) signs or symptoms suggesting an excess of catecholamines, with or without arterial hypertension, (2) arterial hypertension that requires three or more antihypertensive drugs for its management, (3) unexplained blood pressure variability, (4) paradoxical response in surgical interventions after the use of anesthesia and beta adrenergic receptor blockers or (5) in the event of the incidental finding of an adrenal tumor [8, 9].
The preferred diagnosis test is the determination of free metanephrines (normetanephrine and metanephrine) in urine and plasma [2, 5]. Plasma determination is considered superior to the urinary one, with a sensitivity of 97.9% and a specificity of 94.2% [8, 10, 11]. The treatment of pheochromocytoma is based on a multidisciplinary intervention, including the management of symptoms such as hypertension, headache, kidney damage. However, the definitive treatment is surgery to remove the neuroendocrine tumor [5, 8].
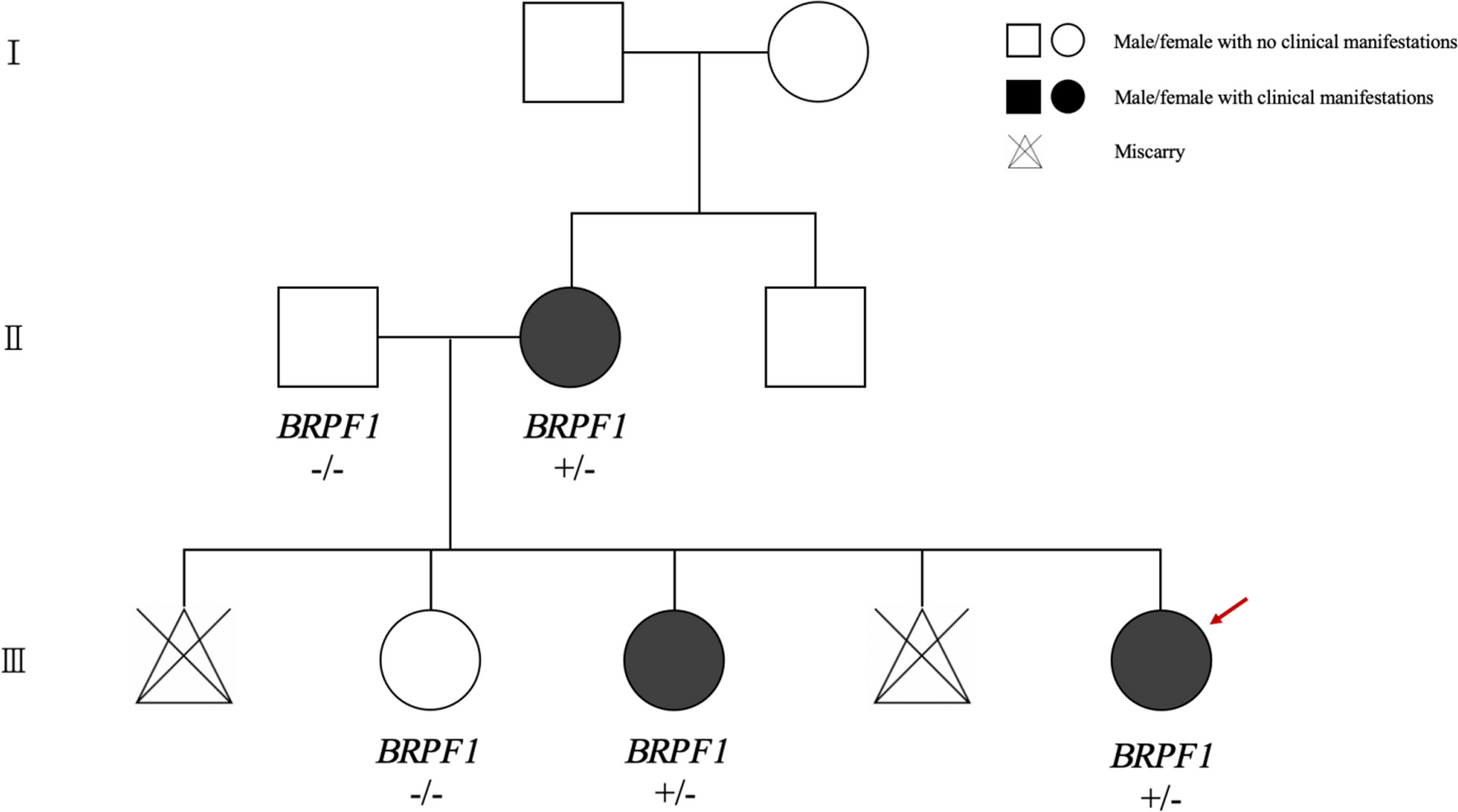
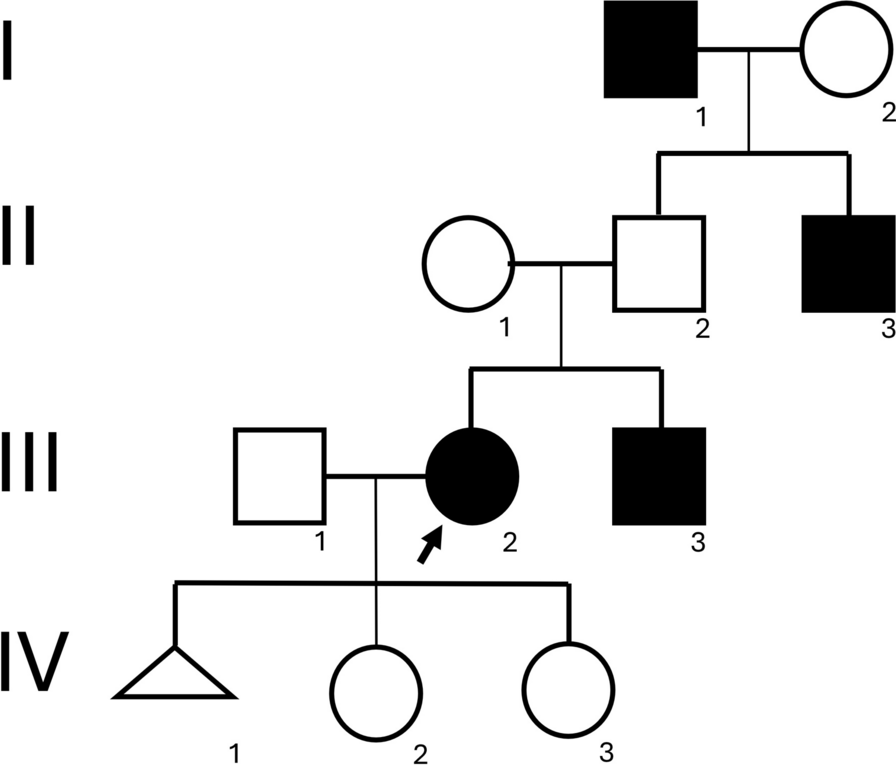
Pheochromocytoma is a rare disease, and its familial presentation is quite uncommon. The aim of this paper is to report a three-generation phenotypical expression of familial pheochromocytoma.

留言 (0)