In this study, we comprehensively analysed the role of the human cell cytomegalovirus (HCMV) pathway in the development of a variety of cancers by means of bioinformatics methods. We found that the HCMV pathway has a wide range of differentially expressed genes in various cancer samples. Immune infiltration analysis demonstrated that immune cells such as T cells and macrophages play important roles in the tumor microenvironment, providing evidence that HCMV is involved in the regulation of tumorigenesis. Furthermore, 23 pathway key genes were identified, which provide potential targets for the development of HCMV-associated targeted therapies. Overall, our study comprehensively resolved the role of HCMV pathway in the development of various cancers, provided new insights into the relationship between HCMV and tumours, and provided a theoretical basis for the treatment of HCMV-associated tumours.
HCMV invasion induces multiple immune responses. Endothelial cells, dendritic cells, natural killer cells, monocytes, and macrophages become activated in the blood and tissues, producing abundant inflammatory mediators including IL-1β, IL-2, IL-6, TNF-α, and chemokines [48,49,50,51]. This process prompts circulating neutrophils to receive signals from antigen-presenting cells at the site of infection and prepare for a further immune response. Concurrently, the activation of local clotting factors also induces platelet aggregation, further enhancing inflammatory processes [52, 53]. Our analysis revealed that differential gene functions across various cancer types, including leukocyte migration, response to abiotic stimulus, and vasculature development regulation, align with the immune responses seen in HCMV invasion. These shared pathways highlight common mechanisms of immune regulation and inflammation critical to both viral infections and cancer progression. Given these findings, it is crucial to explore additional genetic regulatory layers that may influence these processes. DNA methylation, one of the most extensively studied epigenetic modifications, plays a critical role in essential biological processes such as embryonic development, genomic imprinting, and X-chromosome inactivation [54]. Aberrant DNA methylation can alter the cellular microenvironment, affect gene expression patterns, and lead to various pathological conditions, including cancer [55]. Using the MethSurv tool, we explored individual methylation CpG sites in differentially expressed cancer genes [56,57,58]. For instance, in adrenocortical carcinoma (ACC), we found a significant association between the methylation site cg04721825 in the PRKCA gene (HR: 6.626, 95% CI: 1.561–28.119, P: 0.010349507) and disease risk. Similarly, in bladder cancer (BLCA), the methylation site cg09825327 in the TRAF5 gene (HR: 1.682, 95% CI: 1.136–2.49, P: 0.009355414) was significantly associated with disease risk… These results suggest that variations in methylation sites may play a critical role in cancer development, highlighting an important direction for future research. However, the relationship between HCMV and the host immune system is complex, and once HCMV enters the body, it is able to establish latency in undifferentiated hematopoietic progenitor cells in the bone marrow and then reactivate, leading to a recurrence of the disease [51, 59, 60]. Latently infected monocytes disseminate the virus to various organs, and upon their reactivation, the adaptive immune system activates T helper cells and cytotoxic T cells via T cell receptors while accelerating epigenetic events. This further exacerbates the inflammatory role in pathophysiology, which may precipitate multi-organ diseases, heighten disease risk, and even cause death in severe cases [61,62,63,64].
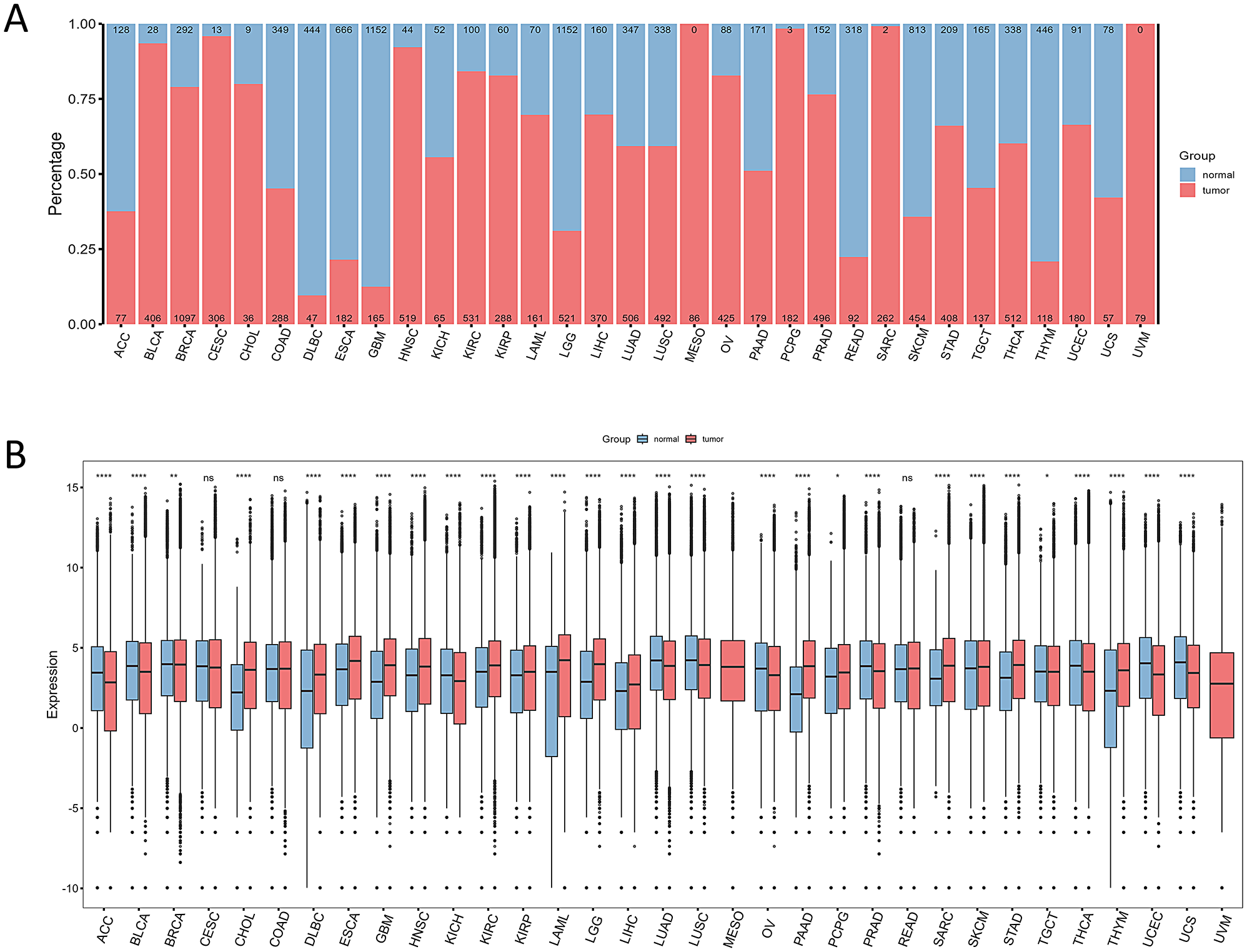
Using the bioinformatics tools CIBERSORT and ESTIMATE, we analyzed the immune cell composition and scoring of 26 cancer types. While the degree and proportion of immune cell infiltration varied across cancers, the infiltration patterns were similar to those of cells involved in innate and adaptive immunity. Fractalkine/CX3C chemokine ligand 1 (CX3CL1) and its receptor CX3CR1 have been found to allow immature dendritic cells to migrate to cancer cells using the expression of their receptor CX3CL1 [65, 66]. The CX3CL1-CX3CR1 axis promotes NK cells to adhere to tumor cells and directly kill cancer cells [67, 68]. Cancers associated with the CX3CL1-CX3CR1 axis include BLCA, STAD, BRCA, GBM, LGG, LIHC, LUAD, LUSC, PAAD, KICH, KIRC, KIRP, OV, PAAD, HNSC, PRAD, TGCT, UCEC, SKCM, etc [69]. . . Results from ESTIMATE revealed high tumor purity across most cancer cohorts, implying a relatively high proportion of tumor cells and low immune activity in the tumor samples. The tumor immune microenvironment represents a highly complex system that is pivotal in driving immunosuppression, distant metastasis, local drug resistance, and response to targeted therapies [70,71,72,73]. Moreover, it is closely related to the clinical prognosis of tumor patients [74]. The tumor microenvironment contains multiple immunosuppressive cell types that are induced by cancer-associated fibroblasts, such as M2 macrophages, regulatory T cells and myeloid-derived suppressor cells. These immunosuppressive cells accumulate abundantly within the tumor immune microenvironment (TIME) and play critical roles in promoting immune evasion and suppression [75,76,77]. , with which our results are also consistent, supporting the hypothesis that enriched immune cells reduce the proportion of tumour cells by enhancing their killing effect on tumour cells and inhibiting their proliferation. We comprehensively analysed the immune infiltration of pan-cancer, which is important for understanding the immune characteristics of tumours and developing corresponding immunotherapy strategies. Future studies can further delve into the interactions of different immune cells in the tumour immune microenvironment to more comprehensively resolve the complexity of the tumour immunoregulatory network.
We identified 25 cancer prognostic genes that are significantly associated with survival (ACC, BLCA, BRCA, CHOL, DLBC, ESCA, GBM, HNSC, KICH, KIRC, KIRP, LAML, LGG, LIHC, LUAD, LUSC, OV, PAAD, PRAD, SKCM, STAD, THCA, THYM, UCEC, UCS), these predictive models of gene composition can effectively distinguish between high and low risk groups of cancer patients. The use of large-scale public databases for biological information mining allows us to efficiently discover these potential cancer prognostic genes [78]. Our findings provide important molecular markers for prognostic assessment and risk stratification of cancer. The study by Anuraga et al. (2021), leveraging multiple database resources such as The Cancer Cell Line Encyclopedia (CCLE) and the Tumor Immune Estimation Resource (TIMER), has successfully identified prognostic biomarkers for breast cancer, contributing valuable insights to the field. Their work not only underscores the efficacy of CCLE in cancer genomics research but also inspired the application of this methodology to pan-cancer analysis, further exploring the commonalities and specificities of molecular characteristics across various cancers [79, 80]. Notably, individual cancer types, such as TGCT, did not show significant prognostic genes in our model, which may be related to the small sample size. In follow-up studies, we will collect more samples to improve statistical efficiency and use experimental techniques to verify the expression and prognostic value of these genes in relevant cancer samples. In addition, we analyzed the mutation spectra of these prognostic genes, and found that about 55% of the genes had mutation information, and the proportion of missense mutations was relatively high. Studies have shown that tumor genomic characteristics, mutation load, and tumor-specific neoantigens are key factors in determining a patient’s response to immune checkpoint blockers, and they may affect the patient’s immunotherapy response. Moreover, tumor mutation load and its associated tumor-specific neoantigens appear to be key ways to predict the potential clinical efficacy of immune checkpoint blockers [81].
The present study identified 23 pivotal hub genes by constructing protein-protein interaction networks and implementing the MCODE algorithm, thus shedding light on the molecular mechanisms underlying the interplay between HCMV infection and host cells. These hub genes were implicated principally in cell chemotaxis and synaptic function modulation, intimating that viruses might harness such processes to facilitate dissemination and proliferation. Meanwhile, promotion of CaM family genes (CALM1, CALM2, CALM3, CALML3, CALML4, CALML5, CALML6) and inhibition of AC family genes (ADCY1, CDCY2, ADCY3, ADCY4, ADCCY5, ADCY6, ADCY7, ADCY8, ADCY9) were observed. These are prognosis-associated genes. Notably, while mutations prevail in AC family genes expressed in manifold cancers (e.g. HNSC, KICH, KIRC, KIRP, LUAD, PRAD), the mutation rates of CaM family genes were mostly 0% across certain expressed cancers (BRCA, LGG, LIHC, PRAD, SKCM, THCA). Additionally, we utilized the GDSC database to screen for drug sensitivities, which provides a comprehensive overview of the correlations between gene expression levels and various drug responses. To further validate these findings, we employed the Connectivity Map (CMap) database, a broad bioinformatics resource used to elucidate the connections between small molecules, biological processes, and disease states by comparing gene expression profiles to predict drug mechanisms, annotate genetic variations, and provide insights for clinical trials [82,83,84]. Interestingly, the results between CMap and GDSC were similar, and more importantly, CMap identified the targets of drug actions. By leveraging CMap, we were able to cross-validate our preliminary findings and gain deeper insights into drug-gene interactions, thus proffering novel theoretical foundations for therapeutic and preventive interventions against the diseases. However, inter-gene interactions are intricate and multifaceted. Whether the pathway hub genes discovered in this study are the major factors influencing HCMV-induced disease progression remains to be further validated. Their purported capacities to exert pivotal roles in diverse HCMV-associated cancers also remain nebulous. Despite unveiling latent molecular mechanisms, inherent limitations exist as a purely in bioinformatical predictive study. Subsequent validation through experimental techniques is imperative to verify the expressional and functional alterations of these genes, alongside their precise roles during HCMV infection. Furthermore, the regulatory mechanisms of the hub genes and their downstream pathways, as well as the influences on viral infection and pathogenic mechanisms of diseases, warrant further in-depth research. Investigations on their consistencies of actions across the gamut of HCMV-induced cancers are necessitated, alongside assessments of their potentials as novel biomarkers or therapeutic targets for diseases. In summary, this study proffered valuable insights into HCMV and host interplay, and invoked multiple scientific issues for scholars across pertinent domains to pursue. We hope that future studies can further uncover the mystery of HCMV mechanism and provide theoretical basis for prevention and treatment of related diseases.


留言 (0)