
記住我
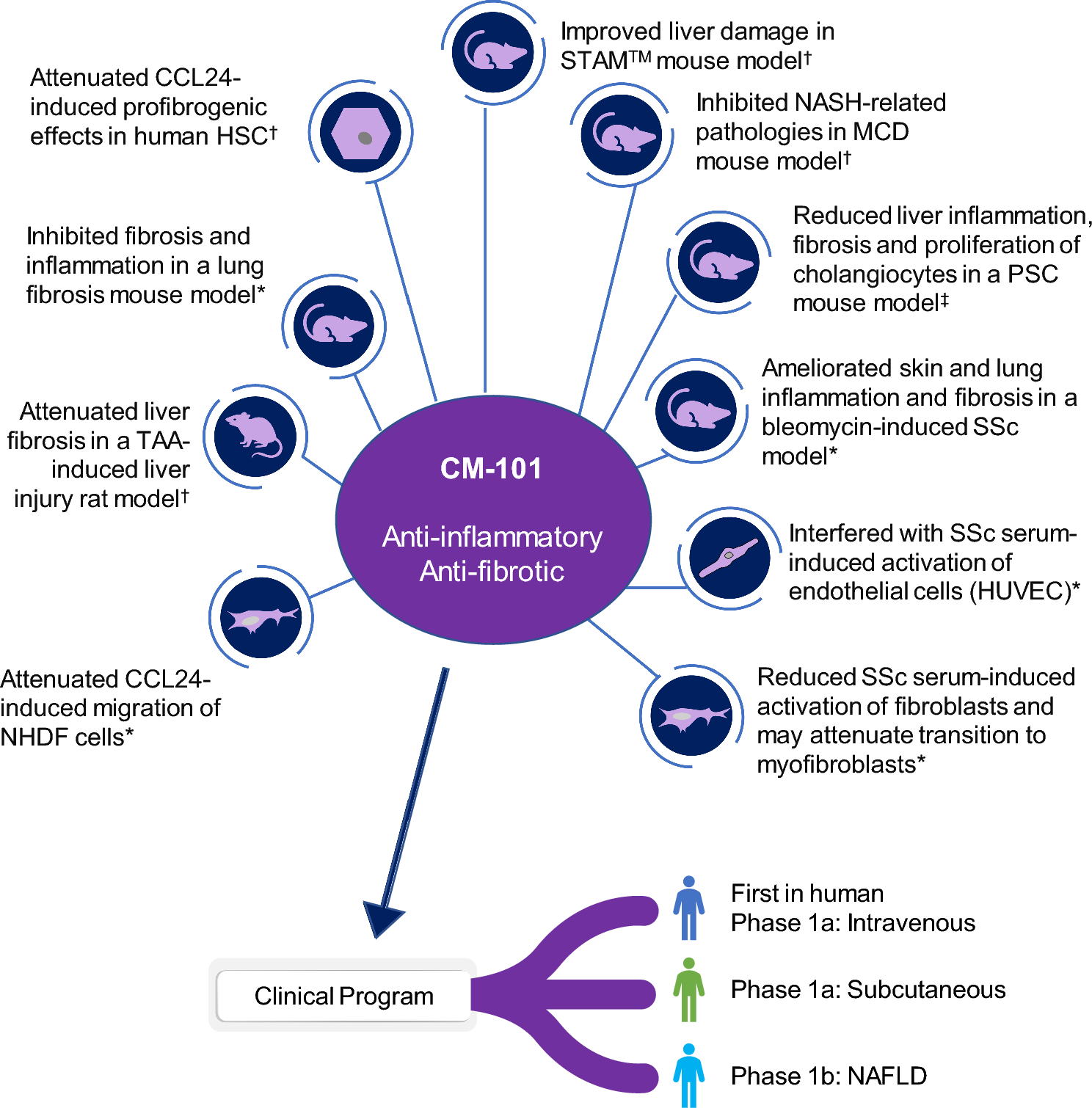


Participants enrolled in the two Phase 1a studies were healthy male volunteers aged 18–45 years who had received no biological treatment with recombinant antibodies, immunological therapy, or anti-cancer treatment. Patients enrolled in the Phase 1b MASLD study were male or female, aged 18–75 years, with normal liver function, and ultrasound-confirmed (FibroScan®; Echosens, Paris, France) MASLD without evidence of MASH. Patients with diabetes type 1 or 2 (if uncontrolled at the time of screening or being treated with insulin), chronic viral hepatitis B or C infection, a history of substance abuse, or significant alcohol consumption (defined as an average >20 g/day in females and >30 g/day in males), or those who had an MASLD activity score of 3 or greater were excluded from the Phase 1b MASLD study. All studies were conducted in accordance with the Good Clinical Practice Guideline as defined by the International Conference on Harmonization and the Declaration of Helsinki. All study participants provided written informed consent prior to any study activity. The protocol, amendments, and informed consent forms for the Phase 1a studies were approved by the Tel Aviv Souraski Medical Center ethics committee; for the Phase 1b MASLD study, informed consent forms, protocol, and amendments were approved by the Hadassah Medical Center institutional review board.
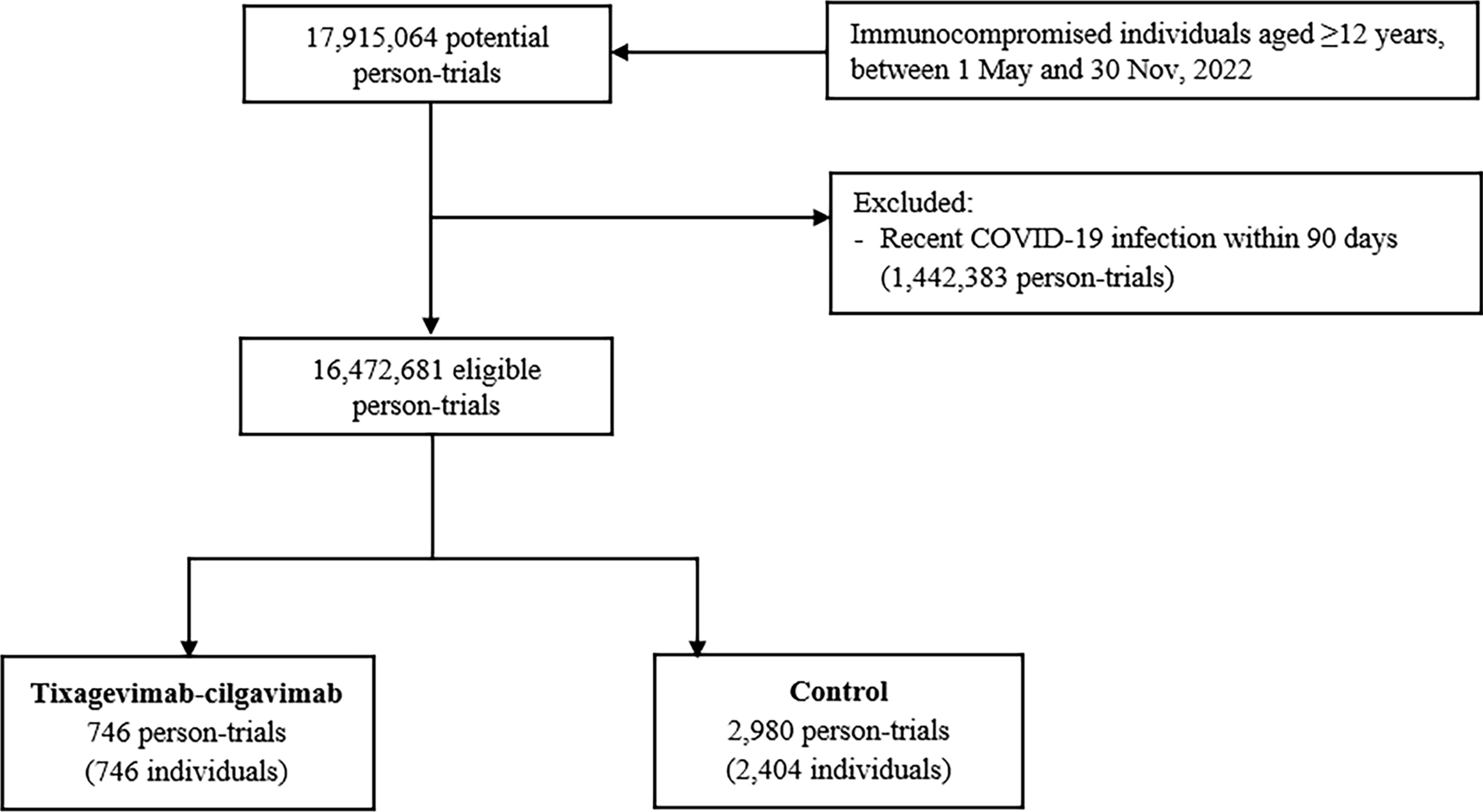
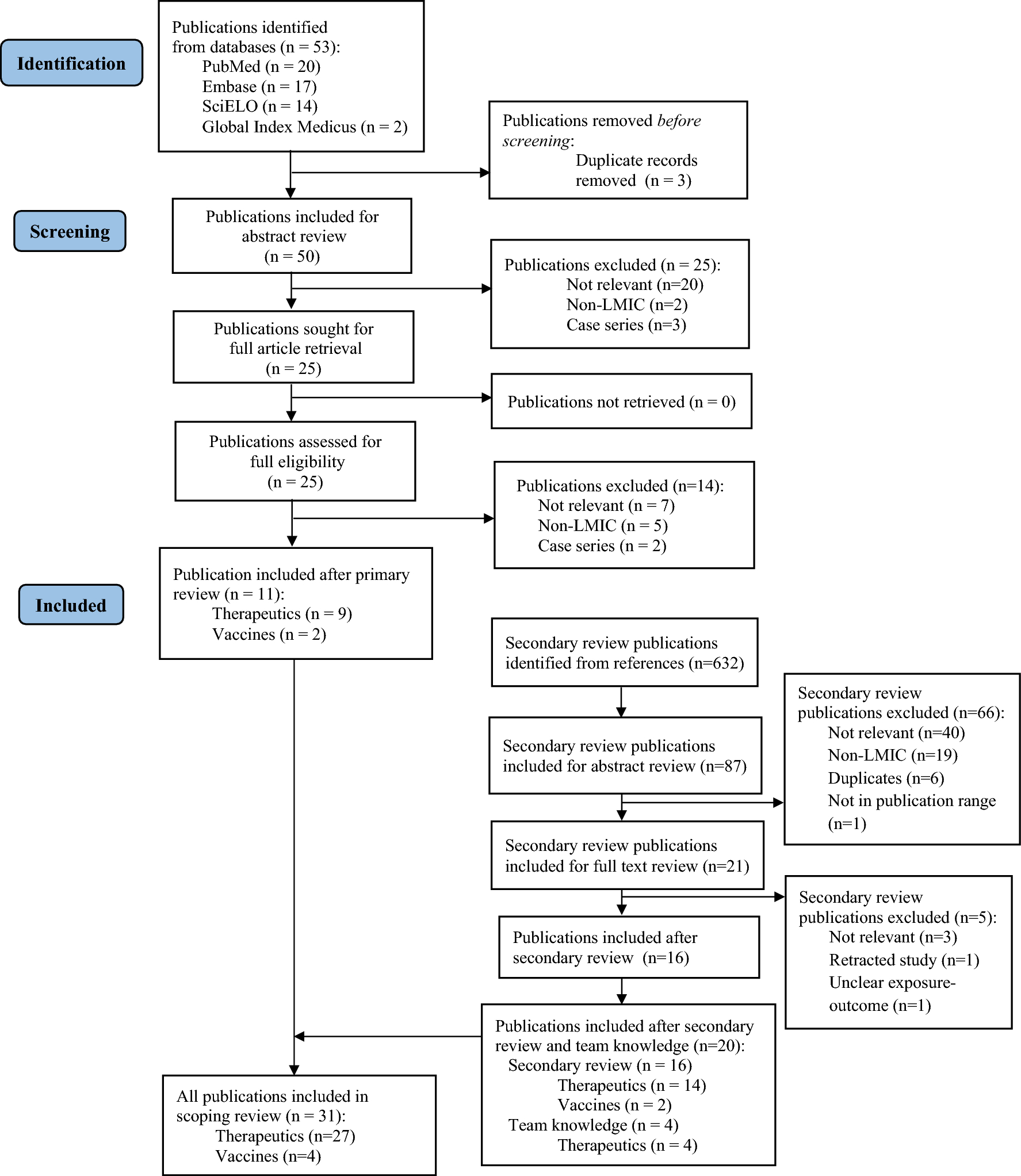
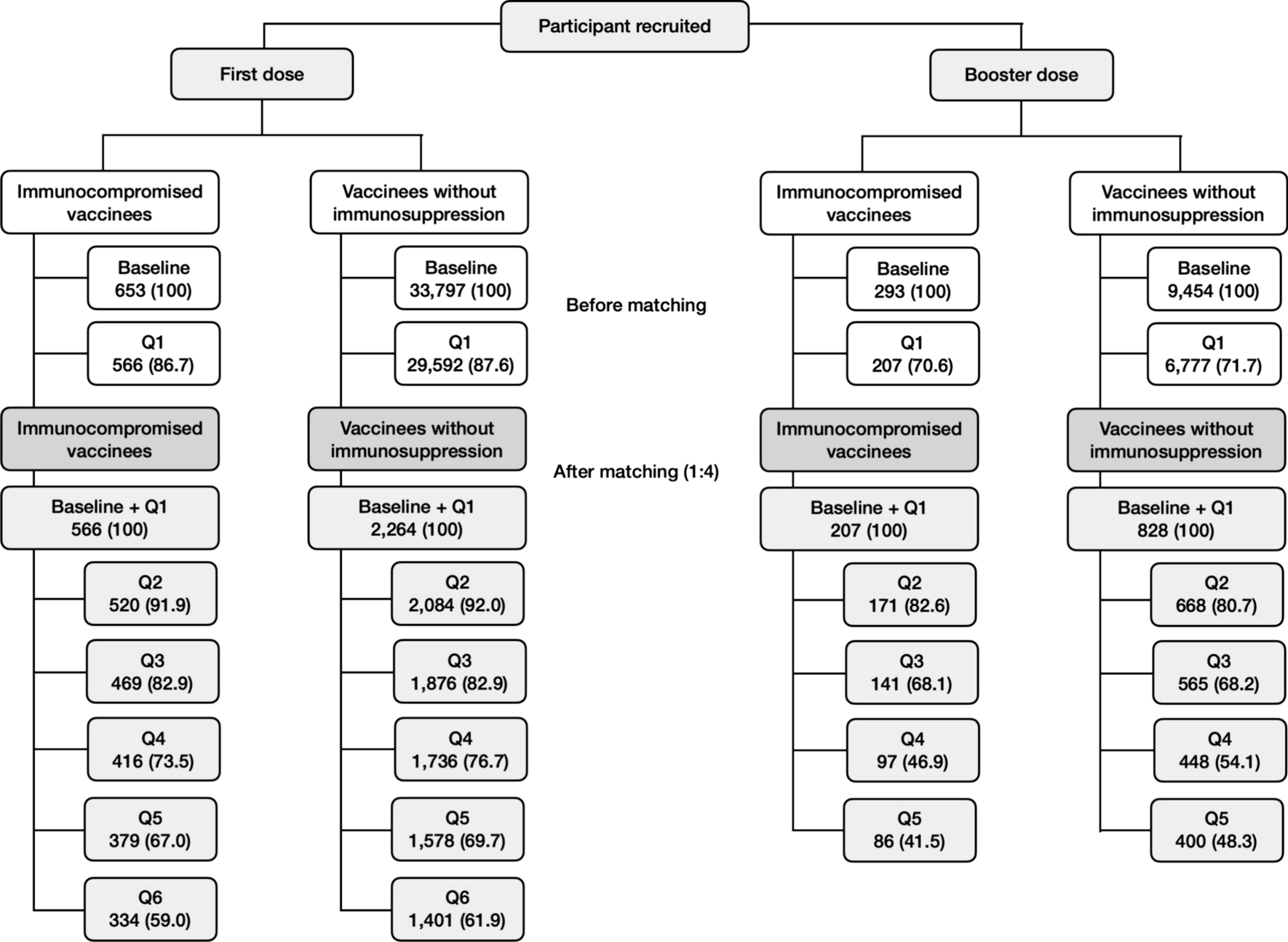
2.2 Study Design and TreatmentThe Phase 1a, first-in-human, single-center, randomized, double-blind, placebo-controlled, single-dose, dose-escalation IV study (Phase 1a IV study) investigated the safety, tolerability, and pharmacokinetics of CM-101 administered via IV infusion. Participants were randomized 3:1 to receive a single dose of either CM-101 or placebo via a 60-min IV infusion within one of four dose groups (0.75 mg/kg, 2.5 mg/kg, 5.0 mg/kg, and 10.0 mg/kg) (Fig. 2). For safety considerations, the first two participants in each dose group—one receiving CM-101 and one receiving placebo—were dosed initially, followed by the remaining participants within the group.
Fig. 2
Study designs for the Phase 1a studies and the Phase 1b study. IV intravenous, MASH metabolic dysfunction-associated steatohepatitis, MASLD metabolic dysfunction-associated steatotic liver disease, SC subcutaneous
The Phase 1a, single-center, randomized, double-blind, placebo-controlled, single-dose SC study (Phase 1a SC study) investigated the safety, tolerability, and pharmacokinetics of CM-101 administered via SC injection. Participants were randomized 3:1 to receive a single SC injection of either CM-101 5.0 mg/kg or placebo in the lower abdomen (Fig. 2).
The Phase 1b MASLD study was a double-blind, randomized, placebo-controlled, single-center, repeated-dose, two-cohort study that investigated the safety, tolerability, and pharmacokinetics of CM-101 in patients with MASLD without evidence of MASH. In cohort 1, patients were randomized 3:1 to receive CM-101 2.5 mg/kg or matching placebo via a 60-min IV infusion once every 3 weeks for 12 weeks (Fig. 2). In cohort 2, patients were randomized 3:1 to receive CM-101 5.0 mg/kg or matching placebo via a 30-sec slow SC injection into the abdominal wall once every 3 weeks for 12 weeks.
2.3 Assessments2.3.1 SafetyThe primary safety assessment for the two Phase 1a studies was the incidence, severity, and duration of adverse events (AEs) following a single dose of CM-101 in healthy volunteers. Adverse events were reported by the participants or observed by the investigator and included changes from baseline in vital sign measurements; electrocardiogram tracings; physical examination findings; concomitant medication concerns; and laboratory assessment findings that included hematology tests, blood chemistry tests, and urinalysis. Participants were monitored for AEs and remained under medical supervision for approximately 24 h post-dose. Adverse events were assessed throughout the entire 42 days of follow-up. Adverse event severity was recorded and graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI-CTCAE), Version 4.03, and coded into the database according to the Medical Dictionary for Regulatory Activities (MedDRA) version 21.0. Causality assessment involved a comprehensive review of factors such as known pharmacological effects, concurrent medications or medical conditions, and any relevant laboratory findings.
The primary safety assessment for the Phase 1b MASLD study was the incidence, severity, and duration of AEs that included clinically significant laboratory abnormalities following administration of CM-101 in patients with MASLD without evidence of MASH throughout 12 weeks of treatment and 6 weeks of follow-up. Results of vital sign measurements, physical examination findings, 12-lead electrocardiogram tracings, laboratory findings on hematology tests, blood chemistry tests, urinalysis, and injection-site reactions were assessed.
A data monitoring board reviewed severe or serious drug-related AEs and determined if events were to be considered dose-limiting toxicities. A dose-limiting toxicity was defined as a clinically significant drug-related AE or abnormal laboratory value assessed as unrelated to disease progression, intercurrent illness, or concomitant medication use and occurring up to 14 days of dosing in the Phase 1a single-dose studies and up to 3 days after receiving any of the first three doses (Day 45) in the Phase 1b MASLD study.
2.3.2 PharmacokineticsIn the Phase 1a studies, fasting blood samples were drawn pre-dose (within 90 min before dosing) and at various predefined time points after IV infusion or SC injection. Total CM-101 blood concentrations (free, partially free, and bound antibody) were quantified using an enzyme-linked immunosorbent assay. Pharmacokinetic analyses of participant concentration versus time data were performed using non-compartmental and compartmental pharmacokinetic approaches. Pharmacokinetic parameters included maximum serum concentration (Cmax), time to maximum serum concentration (Tmax), area under the concentration-time curve from time 0 to time t (AUC0–t), area under the concentration-time curve from time 0 extrapolated to infinity (AUC0–∞), elimination rate constant during the terminal phase (kterminal), terminal elimination half-life (t1/2), apparent volume of distribution during the terminal phase (Vz), apparent volume of distribution at steady state (Vss), and clearance (CL).
In the Phase 1b MASLD study, blood samples were collected before and immediately after each infusion, and at 2, 4 and 6 h after doses 1 (Day 1) and 5 (Day 84). CM-101 serum concentrations were determined using an enzyme-linked immunosorbent-based assay with values below the lower limit of quantification imputed as zero. Compartmental pharmacokinetic analyses were performed; parameters included pre-dose serum concentration (C0), Cmax, Tmax, area under the serum concentration-time curve from time of administration up to the last time point with a measurable concentration post-dosing (AUClast), AUC0−∞, elimination rate constant (λz), and t1/2.
2.3.3 PharmacodynamicsAn exploratory objective for all three studies was to investigate the effect of different doses on serum CCL24 levels pre- and post-treatment. Serum samples were collected pre-dose and at various predefined time points after IV infusion or SC injection. A human CCL24/Eotaxin-2/MPIF-2 Quantikine ELISA kit (DCC240B; R&D Systems, Minneapolis, MN, USA) was used to quantitate levels of CCL24 across time points.
In the Phase 1b MASLD study, the effect of CM-101 on fibrotic process was evaluated using serum fibrotic and collagen turnover biomarkers. The change from baseline at Week 15 (after five doses) in tissue inhibitor of metalloproteinase-1 (TIMP1), tissue inhibitor of metalloproteinase-2 (TIMP2), platelet-derived growth factor AA (PDGF-AA), N-terminal type III collagen propeptide (Pro-C3), N-terminal type IV collagen propeptide (Pro-C4) and type III collagen fragment (C3M) was calculated. Serum levels of soluble fibrotic biomarkers, such as TIMP1, TIMP2, and PDGF-AA were tested using Milliplex® (Merck KGaA, Darmstadt, Germany) multiplex analysis. Serum levels of Pro-C3, Pro-C4, and C3M were analyzed by a contract laboratory (Nordic Bioscience Laboratory, Copenhagen, Denmark) according to their protocols.
2.4 Statistical AnalysisAll measured variables and derived parameters were tabulated by descriptive statistics. For categorical variables, summary tables were provided giving sample size and absolute and relative frequency by individual treatment (CM-101 or placebo). For continuous variables, summary tables were provided giving sample size; arithmetic mean; standard deviation; and minimum, median, and maximum values by individual treatment (CM-101 or placebo). Data were analyzed using SAS software version 9.3 or higher (SAS Institute, Cary, NC, USA). No formal sample size calculation was performed for these studies, as they were designed to assess preliminary safety and pharmacokinetic data. No data imputation for missing data was applied. In the Phase 1a studies, non-compartmental and compartmental pharmacokinetic analyses were performed using PK Solver version 2.0 software [7] validated with SAS version 9.4 (SAS Institute) and STATA SE version 13.0 (StataCorp, LLC, College Station, TX, USA). In the Phase 1b MASLD study, compartmental pharmacokinetic analyses were performed using MONOLIX 2018 R2 software (Lixoft, Antony, France).
留言 (0)