
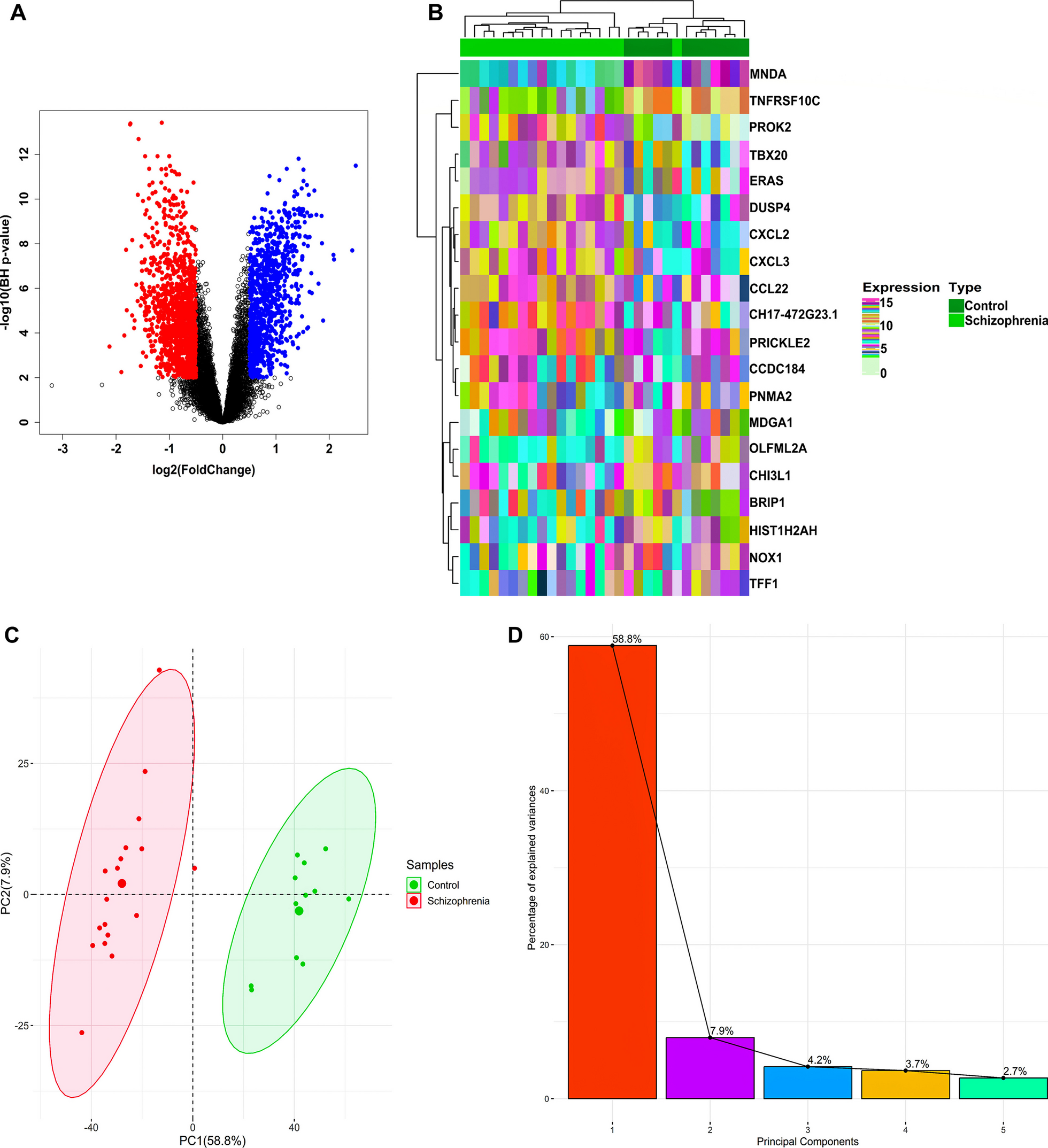
記住我
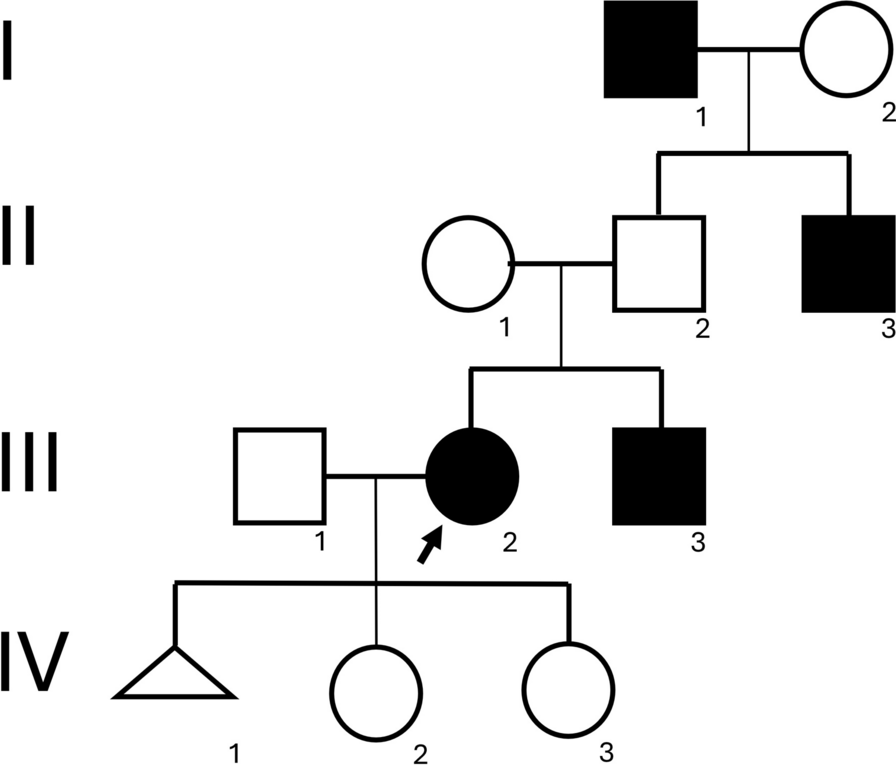
UniProt (https://www.uniprot.org) [17] database was used to extract the amino acid sequence of α-synuclein having Accession No. AAA16117.1. ProtParam (https:// web.expasy.org/protparam) [19] tool predicted different chemical and physical properties of the α-synuclein. It consists of 140 amino acid residues, predicted molecular mass of 14.5 kDa, predicted pI is 4.67, instability index of 25.47 means it is stable at room temperature, estimated half-life is greater than 20 h, aliphatic index is 69.64 that shows this protein is thermodynamically stable over a broad range of temperature and it also contains high amount of hydrophobic amino acid residues as predicted by its, grand average of hydropathicity (GRAVY) which is -0.403. The SMART (http://smart.embl-heidelberg.de) [20] web server predicted that the α-synuclein protein has no specific domains depending upon the sequence of amino acids. NetPhos 3.1 (https://services.healthtech.dtu.dk/service.php?NetPhos-3.1) [21, 22] and GPS 3.0 (http://gps.biocuckoo.org/online.php) [23] server anticipated phosphorylation of residues S9, T33, Y39, S42, T54, T59, T81, T92, Y125, S129, and Y133 in α-synuclein protein.
According to the subcellular location findings by TargetP 2.0 (https://services.healthtech.dtu.dk/service.php?TargetP-2.0) [24] and SignalP 5.0 (https://services.healthtech.dtu.dk/service.php?SignalP-5.0) [24] α-Synuclein is a secretory protein and has no signal peptide. TOPCONS (https://topcons.cbr.su.se) [25] showed that α-synuclein is not a homologous transmembrane protein.
STRING (https://string-db.org) [26] database identified both known and potential interacting partners of α-synuclein. They were HSPA8 (Heat shock protein member 8), TH, VAMP2 (Vesicle-associated membrane protein 2), PARK7 (Parkinson disease protein 7), TPPP (Tubulin polymerization-promoting protein), PARK2 (Parkin), HSPA4 (Heat shock 70 kDa protein 4), SLC6A3 (dopamine active transporter), SNCAIP (Synphilin-1 also known as synuclein, alpha interacting protein), and LRRK2 (Leucine-rich repeat kinase 2) as shown in Fig. 1.
Fig. 1
Possible physical and functional interacting partners of α-synuclein identified by STRING database
Binding pocket analysisCASTp was used to find probable binding sites and the binding site residues of α-synuclein. The best predicted pocket is composed of total 13 amino acid residues, whose solvent accessible surface area and pocket volume are 148.314 Å2 and 599.861 Å3, respectively, as shown in Table 2.
Table 2 Potential binding site of α-synuclein anticipated by CastPDeleterious nsSNPs prediction in coding regionEnsembl genome browser (https://asia.ensembl.org/index.html) [18] was used to collect the nsSNP data for α-synuclein. There were total 184 numbers of nsSNPs present in the sequence which were missense variants, out of which 10 were in the splice region. We employed various in silico tools to predict deleterious nsSNPs from 174 missense variants in the coding region.
Classification and evaluation of predicted deleterious nsSNPs by various in silico toolsIn present study, I-Mutant 3.0(http://gpcr2.biocomp.unibo.it/cgi/predictors/I-Mutant3.0/I-Mutant3.0.cgi) [28] identified 88 nsSNPs with DDG value < 0 which may reduce protein stability. The DDG value is calculated from the unfolding Gibbs free energy value of the mutated protein minus the unfolding Gibbs free energy value of the wild type (Kcal/mol), PolyPhen-2(http://genetics.bwh.harvard.edu/pph2) [29] predicted 79 nsSNPs as probably damaging, PhD-SNP(https://snps.biofold.org/phd-snp/phd-snp.html) [28] with a score of > 0.5 predicted 25 nsSNPs to be disease causing, PANTHER (http://www.pantherdb.org/tools/csnpScoreForm.jsp) [30] identified 40 nsSNPs as probably damaging, SNPs&GO (https://snps.biofold.org/snps-and-go/snps-and-go.html) [31] predicted 87 nsSNPs as disease causing with a score of > 0.5 and PROVEAN (http://provean.jcvi.org/seq_submit.php) [32] anticipated 60 nsSNPs with a score of ≤ -2.5 as deleterious as shown in Fig. 2.
Fig. 2
Identification and classification of deleterious nsSNPs in α-synuclein: (A) Flowchart describing the in silico procedure for evaluating deleterious nsSNPs and subsequent structural analysis (B) Bar diagram representing the number of nsSNPs predicted as deleterious by following in silico tools: I-Mutant 3.0 (88 nsSNPs), PolyPhen-2 (79 nsSNPs), PhD-SNP (25 nsSNPs), PANTHER (40 nsSNPs), SNPs&GO (87 nsSNPs), PROVEAN (60 nsSNPs)
Out of all these nsSNPs, three were found to be damaging by all in silico tools used. They are rs1433622151 (G25S), rs1261243630 (V66E), and rs745815563 (V77D) found as damaging by I-Mutant 3.0, PolyPhen-2, PhD-SNP, PANTHER, SNPs&GO, PROVEAN which are located in the predicted binding pocket. To determine the possible deleterious effects of these mutations, we used five additional in silico tools: MUpro, INPS, DUET, SDM, and mCSM. Tables 3 and 4(a) show the result. We also computed physicochemical properties of these 3 nsSNPs using ProtParam (https:// web.expasy.org/protparam) [19] as shown in Table 4(b).
Table 3 Deleterious nsSNPs identified in α-synuclein by various in silico toolsTable 4 (a) Impact of prioritised nsSNPs of α-Synuclein on its structure; (b) Impact of prioritised nsSNPs of α-Synuclein on its predicted physical and chemical propertiesGeneration of in silico mutantsThe mutation wizard of PyMOL was used in our study to genarate an in silico mutant structures of variations as shown in Table 5, that had been found deleterious by all in silico tools and are also located in predicted binding pocket. The variants are—rs1433622151 (G25S), rs1261243630 (V66E), and rs745815563 (V77D).
Table 5 In silico mutant structures of prioritised variantsMolecular Dynamic (MD) SimulationWe performed MD simulations of 100 ns for wild-type α-synuclein and its mutant structures to further comprehend the structure and functional properties of the predicted disease-causing mutations. After the end of the MD simulations, the structural integrity and stability of wild-type and mutant structures was determined using the various analyses like root-mean-square deviation (RMSD), root-mean-square fluctuations (RMSF), radius of gyration (Rg), and solvent accessible surface areas (SASA). Effect of mutants on the helix properties of protein was also studied. Essential dynamics based on principal component analysis of wild-type and mutant α-synuclein structures were also used to study and understand biologically relevant motions.
The RMSD approach was used to evaluate the protein’s structural stability and its mutations on a consistent time frame [44]. RMSD assessment was carried out on all mutant structures as well as the wild type of α-synuclein. In both wild-type α-synuclein and its mutant structures, structural variations have been detected. Compared to the wild-type and the mutant structures, all three mutant structures showed significant deviation from wild-type α-synuclein as shown in Fig. 3a and Table 6. The average values of RMSD for wild type, G25S, V66E, and V77D were 3.28 nm, 2.60 nm, 2.90 nm, and 3.03 nm, respectively. The average of all the three mutations indicate that these variants alter the protein structure of α-synuclein. We used the RMSF to check if the amino acid alteration changed the dynamic characteristics of the residues. The flexible and non-flexible portions of wild-type and mutant α-synuclein structures can be identified using the RMSF parameter. In this analysis, we found fluctuations in protein structure with all the three mutations throughout the simulation up to 100 ns. When compared to wild-type structures, the V66E mutant structure displayed more fluctuations as shown in Fig. 3b and Table 6. The average RMSF values for wild type, G25S, V66E, and V77D were 0.66 nm, 1.15 nm, 1.51 nm, and 0.85 nm, respectively. The total RMSF data revealed that all three mutations show the increase in the fluctuations as compared to the wild-type structure of the protein. The radius of gyration (Rg) is explained as the mass-weight root-mean-square distance between a cluster of atoms and their shared centre of mass. As a result, it gives data on the protein’s overall dimension. In this study, we found that the Rg value for G25S was higher as compared to V66E and V77D as shown in Fig. 3c and Table 6. The average Rg values for wild type, G25S, V66E, and V77D were 2.61 nm, 4.06 nm, 2.99 nm, and 2.59 nm, respectively. The Rg findings suggest that though G25S shows higher values, all three mutations influence folding pattern of wild-type structure. We applied SASA to investigate wild-type and mutant α-synuclein structures to determine the solvent accessibility of α-synuclein and the implications of mutations on the solvent effect of α-synuclein as shown in Fig. 3d and Table 6. The average SASA values for wild type, G25S, V66E, and V77D were 115.24 nm2, 128.14nm2, 128.91nm2, and 118.7nm2, respectively. The SASA values of mutant structures are higher than native form revealed that due to the influence of mutations structural changes occurred.
Fig. 3
Molecular dynamics simulation results of the wild-type α-synuclein and 3 mutant (V77D, V66E, and G25S) structures of α-synuclein. (a) Root-mean-square deviation, (b) Root-mean-square fluctuation, (c) Radius of gyration and (d) Solvent accessible surface area. Wild type is represented in black, mutant G25S in red, mutant V66E in green, and mutant V77D in blue
Table 6 Time averaged structural properties obtained from MD simulations of native and mutant (G25S, V66E, and V77D) structures of α-synucleinThe essential dynamics (ED) approach was utilised to capture the fundamental, biologically relevant motions from the global trajectories of wild-type and mutant α-synuclein structures for the principal component analysis. In ED, two variables, PC1 (Principal Component 1) and PC2 (Principal Component 2), measure the aggregate fluctuations of the most variable areas of α-synuclein. The eigenvalue ranks and two-dimensional projections of the significant variations are depicted in Fig. 4. The initial conformations of wild type and mutants are reported by blue dots, whereas the intermediate and final conformations are represented by the white and red dots, respectively. The continuous colour scale from blue to white to red dots indicates that there were periodic jumps between these conformers throughout the trajectory. The projections implied that, in comparison with the wild-type α-synuclein, the mutants show distinct conformational changes in the essential dynamics of α-synuclein. The initial conformations of wild-type α-synuclein protein are clustered in left and right top side of essential subspace while the final conformations are tightly clustered in bottom right of essential subspace. The conformational subspace occupied by wild-type α-synuclein along PC1 is 67.14% and 7.66% for PC2. Further, the conformational subspace occupied mutant G25S along PC1 is 37.36% and 23.58% for PC2. Similarly, the conformational subspace occupied by mutant V66E along PC1 is 56.66% and 20.94% for PC2. Lastly, the conformational subspace occupied by mutant V77D along PC1 is 54.81% and 12.74% for PC2. By PCA graphs, we found that all mutants show higher flexibility than wild type as shown in Fig. 4. The total PCA analysis revealed that all mutants affect the stability of α-synuclein protein, which line with the observations of all the initial analysis. Moreover, from the PCA analysis, it has been observed that the trace of covariance matrix values is altered in comparison with native form which again indicated that missense mutations are significantly alter the conformational behaviour of alpha synuclein protein.
Fig. 4
Principal component (essential dynamics) analysis of the wild-type α-synuclein and mutant G25S, V66E, and V77D structures of α-synuclein. (a) wild-type α-synuclein, (b) G25S mutant, (c) V66E mutant and (d) V77D mutant
We also calculated variations caused in helix properties of structure of the wild type and mutants of α-synuclein by using gmx helix module of GROMACS. In this analysis, various helical properties like helix radius, twist, rise per residue, total helix length, RMS deviation from ideal helix, average phi-psi angles, helicity per residue, and average Calpha—Calpha dihedral angle were computed to understand the effect of the mutation on the wild-type α-synuclein protein. The various values obtained by these analysis are shown in Table 7. All helix properties showed alterations in all mutants as compared to wild-type α-synuclein.The graphs showing the variations in different helix properties are given in Additional file 1.
Table 7 Average values of various helix properties of wild-type α-synuclein and mutant structuresOverall molecular dynamics simulation results show that the mutations affect the structural stability and dynamic behaviour of the α-synuclein protein. Therefore, using a molecular dynamics simulation we compared the structures of wild type and all three mutants of α-synuclein protein in this study. Throughout the 100 ns simulation, structural alterations were seen in all three mutants of the alpha-synuclein protein as compared to the wild type. These nsSNPs of the α-synuclein protein may destabilise its structure, which may have an impact on its function, according to this molecular dynamics study. If the activity of α-synuclein protein is disrupted, its cellular functioning may be compromised, leading to serious human malfunctions such neurodegenerative illnesses.
留言 (0)