
記住我
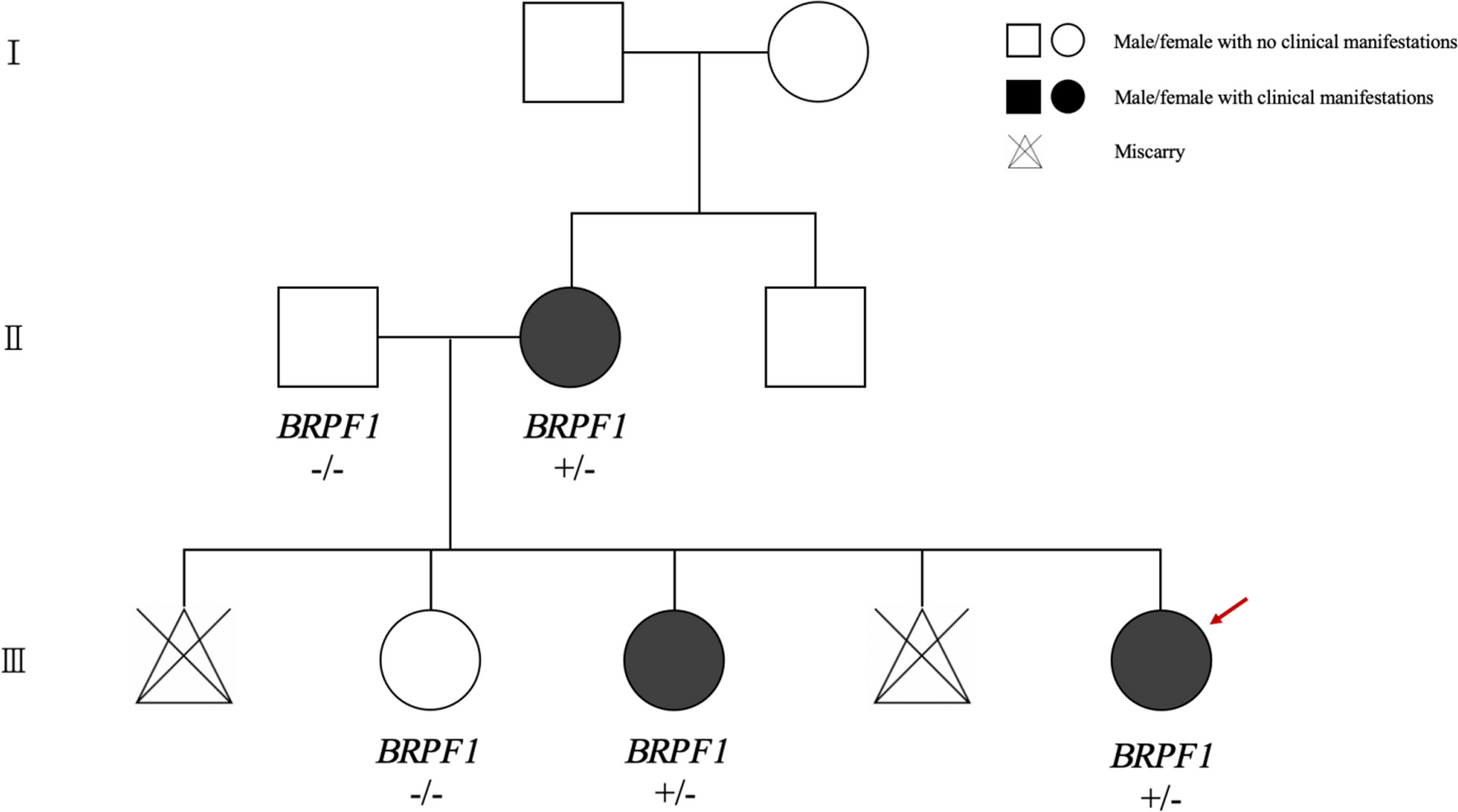
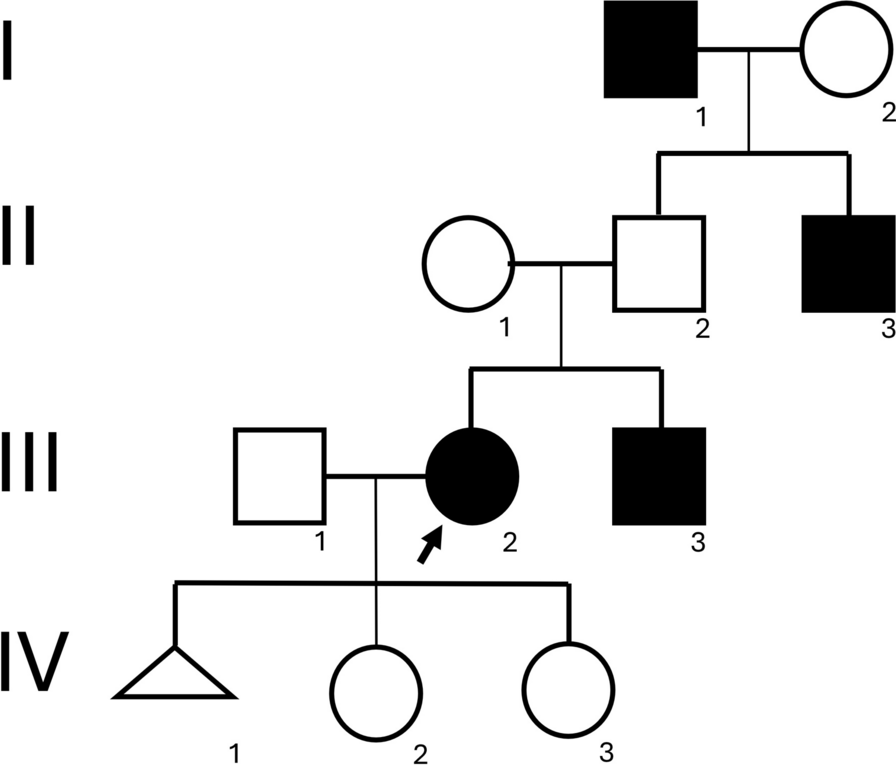
The patients are cousins from a multiply consanguineous Syrian family (Fig. 1). They presented to The Human Genetics Department in November 2022; and they have been thoroughly investigated by us for the past 10 months. Each of the family members underwent a work-up including a thorough clinical evaluation. and routine blood tests (complete blood count, serum electrolytes, blood glucose levels, cholesterol, thyroid, liver and renal function tests, and phosphatase alkaline levels). Extensive genetic testing was further done. Magnetic Resonance Imaging (MRI) was carried out following the institution’s standard clinical protocols, which typically involved acquiring axial, coronal, and sagittal Spin Echo T1 HF, Spin Echo T1, FSE T2, Fast FLAIR T2.
Fig. 1 Isolation of genomic DNA
Isolation of genomic DNAConsent forms were collected from all family members. DNA was extracted from Leucocytes by standard salt-precipitation methods.
Whole exome sequencingWhole exome sequencing (WES) was carried out on two affected individuals (V6 and V7) at 3billion Inc. Illumina NovaSeq 6000 system (San Diego, CA, USA) was used for sequencing as 150bp paired-end reads. Alignment to the GRCh37 human reference genome was done using BWA-MEM2, and samtools v1.15 was used for bam file sorting and marking duplicates [10, 11]. Recalibration and variant calling for single nucleotide variants (SNVs) and small insertion/deletion variants (indels) were performed using GATK v4.2 [12]. Structural variants (SVs) were called using CoNIFER 0.2.2v, and 3bCNV, an internally developed tool [13]. Variants were annotated, filtered, and classified using EVIDENCE v3.2 which incorporates Ensembl Variant Effect Predictor (VEP) for annotation and the American College of Medical Genetics and Genomics (ACMG) guideline for classification [14]. The filtered and classified variants were manually reviewed by medical geneticists and physicians. The most likely variants that can explain the patient’s phenotype were selected for reporting.
Sanger sequencingThe selected variant was studied in the patients V2 and V5 by Sanger sequencing. Genomic and cDNA sequences of ADGRG1 were obtained from UCSC Genomic Browser (NM_201525.4). Primers used for PCR amplification were designed using Primer3 software (http://frodo.wi.mit.edu) to amplify the region surrounding the mutation detected by exome sequencing. PCR products were purified by exonuclease I/Shrimp Alkaline Phosphatase treatment (ExoSAP-IT; Fisher Scientific SAS, Illkirch, France) according to the manufacturer’s instructions and both strands were sequenced using the Big Dye® Terminator v1.1 Cycle Sequencing Kit (Applied Biosystems). Sequence reactions were purified on Sephadex G50 (Amersham Pharmacia Biotech, Foster City, CA) and capillary electrophoresis was performed on Genetic Analyser 3100 (Applied Biosystems). Electropherograms were analyzed on the Sequence Analysis Software version 5.2 (Applied Biosystems) and aligned with the wild-type SAMD9 gene sequence using ChromasPro version 1.22 (Technelysium, Queensland, Australia).
Clinical presentationThe family investigated in this study included 5 affected members (Fig. 1) born after normal vaginal deliveries without any exposure to pre- or perinatal environmental toxins. Psychomotor developmental delay was evident in all the patients in the first few months of life Indeed, limited social interaction, minimal verbal abilities without forming coherent words, inability to achieve toilet training and independent eating were observed in almost all cases. Individual V5 was the only sibling able to socially interact with his surroundings and to say a few words at the age of 4. Siblings V6 and V7 both suffered from oromotor dyspraxia. Patients V3, V5, V7 were not able to walk, and patients V2 and V6 started walking at 4 and 5 years of age, respectively. No dysmorphic features were noted in any of the patients. Microcephaly was noted in cases V6 and V7 (Table 1).
Table 1 Clinical features of the patientsNeurological examination revealed prominent ataxia, pyramidal signs, increased deep tendon reflexes and spasticity for V6 and V7; whereas, mild ataxia was only detected in patient V2. Moreover, individuals V5, V6 and V7 showed tone abnormalities. As for the oculomotor findings, strabismus was present in all individuals; and nystagmus was also detected in patients V6 and V7. Three patients (V5, V6 and V7) suffered from seizure episodes that were noted as early as the first couple of months for patients V6 and V7, or at age of 2 for patient V5. These were not resolved by medications and were still manifesting every 2 or 3 days in the mentioned cases (Table 1).
Imaging findingsA brain MRI performed for V2 at 1 year of age showed subcortical hyperintense signals with bilateral frontal–parietal and occipital topography, in addition to hyperintense zones in the periventricular and frontal horns areas. This was initially thought to be resulting from an infection with Herpes Simplex or Cytomegalovirus (CMV). Another brain MRI performed in this patient at age of 3 did not show any abnormality.
On the other hand, an MRI performed on the patient V5 at the age of 4 years showed the presence of a thick cortex of both cerebral hemispheres with sparse cortical sulci; multiple small hyperintense signal areas in the periventricular and deep white matter of both cerebral hemispheres. The lateral and third ventricles were mildly dilated, with normal fourth ventricles. The basal cisterns in both supra and infratentorial regions were prominent. These findings were first interpreted as being consistent with pachygyria–agyria complex (lissencephaly) (Fig. 2a).
Fig. 2
a Axial T2 brain MRI of patient V5 displaying diffuse high signal intensity on T2 in the white matter alongside posterior polymicrogyria and pachygyria. b Axial FLAIR brain MRI of patient V6 revealing pachygyria and lissencephaly
MRI done on patient V6 at 1 year of age showed hyperintense signals in the periventricular and subcortical areas, accompanied by cerebral atrophy and secondary dilatation of the ventricular system. This was interpreted as lissencephaly (Fig. 2b).
The MRI of V7 performed at 4 years of age revealed an intraventricular cyst bulging into the lateral ventricles' posterior body. It was located inferior to the fornices which were displaced superiorly. There was no solid component, no calcifications, and the content followed CSF on all sequences. The occipital and temporal horns were prominent on the left, suggesting an impairment of the CSF flow caused by the cysts.
Genetic analysisWhole exome sequencing was performed on patients V6 and V7. DNA was sequenced at 3billion using 3B-EXOME proband. A homozygous missense variation in the ADGRG1 gene (NM_201525.4: c.308T > C; p.Leu103Pro) was detected in both (Fig. 3). This variant was not observed in the gnomAD v2.1.1 dataset. Segregation analysis confirmed the presence of this variant in V6, and V7. In silico prediction tools suggest a damaging effect of the variant on the gene product (REVEL: 0.32; 3Cnet: 0.90).
Fig. 3
Electropherogram showing the pathogenic variant identified at homozygous state in patient V6
留言 (0)