
記住我
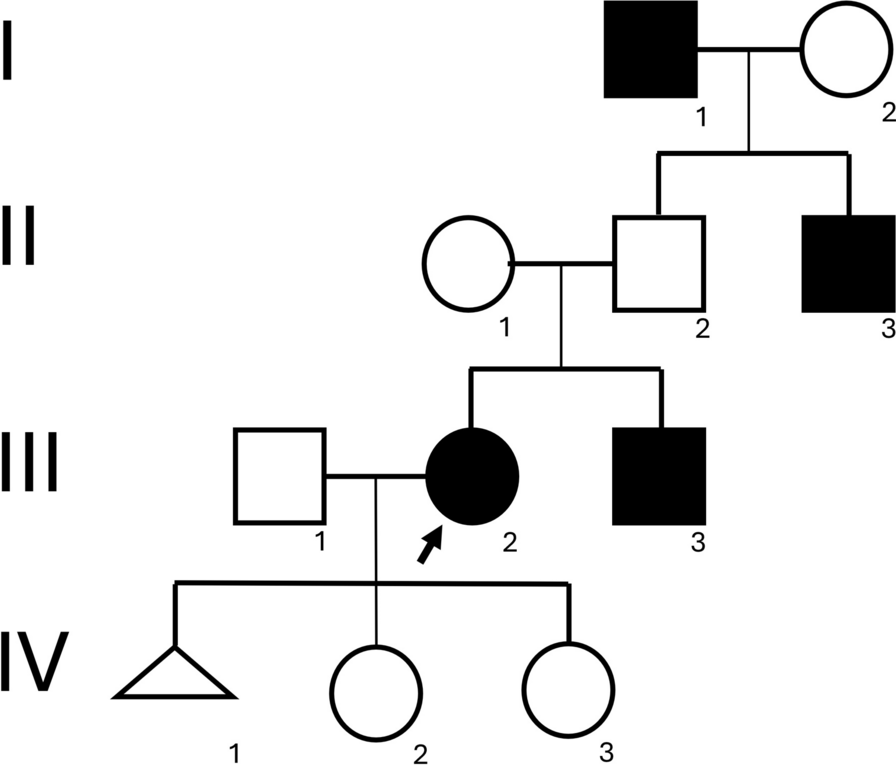
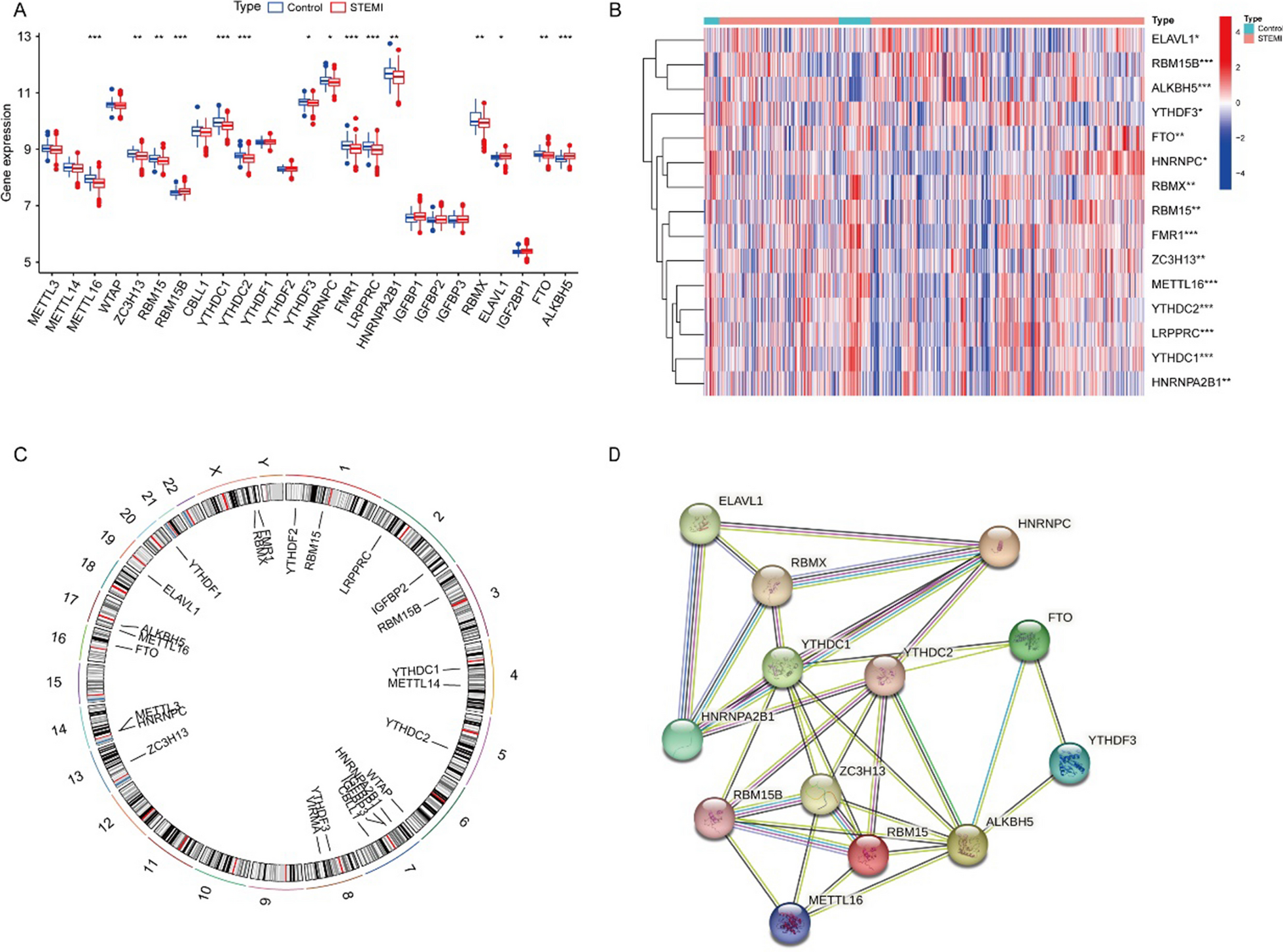
The phytochemical constituents of PVL have been explored in some varieties of PVL influenced by distributed area of origin, environmental conditions, or method of extractions [17, 20, 35]. Different studies reported the phytochemical content of PVL, such as saponin, anthocyanins, flavonols, phenolic acids, proanthocyanidins, and other bioactive compounds, as well as its evidence for anti-inflammatory, anticancer, and immunomodulatory agent [17, 20, 36, 37] as summarized in Table 2. The compound with the highest binding affinity will be selected for molecular dynamic simulations analysis as shown in Fig. 5.
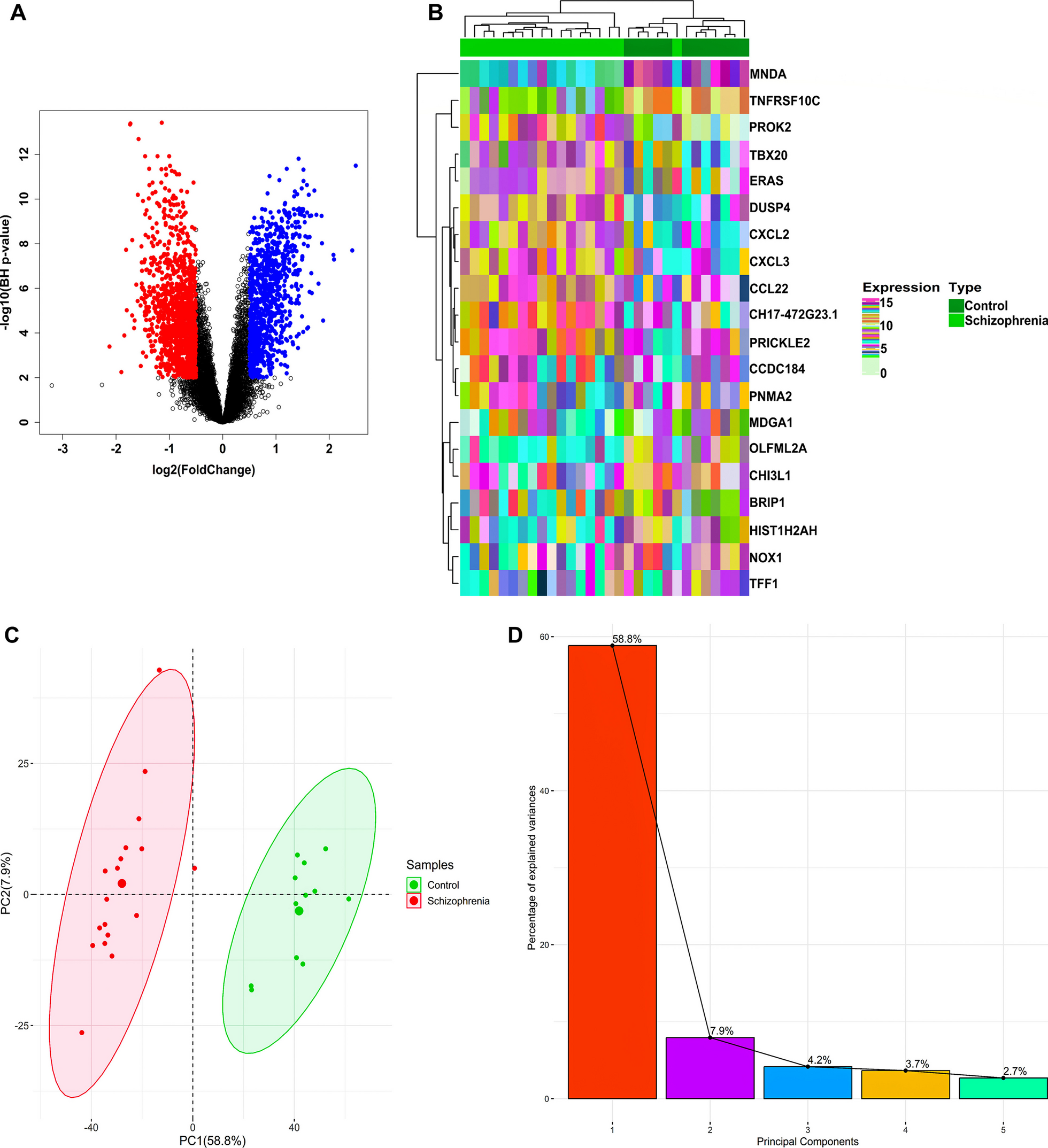
Druglikeness, and ADME prediction toxicity prediction of phytochemical constituents of P. vulgaris LTo know the prediction of pharmacokinetics for each chemical component of PVL, we analyzed the Rule of Five (RoF) score. PVL active compounds were retrieved in sdf file format from the PubChem database (http://pubchem.ncbi.nlm.nih.gov/). Our study revealed that quercetin, kaempferol, myricetin, catechin, 3,4-dihydroxybenzoic acid, and daidzin contained in PVL met the RoF criteria. Besides, other compounds did not meet the RoF criteria due to some factors such as MW > 500, HBA > 10, and LogP > 5, as shown in Fig. 1A and Table 3. RoF is a criterion for evaluating the similarity of a compound to a drug. The requirements for RoF are that the hydrogen bond acceptor (HBA) must be less than 10, the hydrogen bond donor (HBD) must be less than 5, the molecular weight (MW) must be less than 500 g/mol, the H2O partition coefficient (LogP) must be less than 5, and the molar refractivity must be between 40 and 130. The compounds that meet the RoF are predicted to have drug-like properties [38].
Pharmacokinetic properties, including absorption, distribution, metabolism, and excretion, were analyzed among the active compounds of PVL, as shown in Table 3. In absorption, solubility and permeability are essential drug-specific physicochemical properties related to the ability of the drug across the membrane to reach the desired concentration in the systemic circulation [39]. Solubility is critical in developing oral drugs, as low solubility can affect intestinal absorption through the portal circulation. Notably, all PVL compounds have low water solubility (< 0.4 mol/L), indicating poor water solubility related to lipophilicity since they have LogP less than 5 [40]. Low water solubility can lead to a poor dissolution rate and potentially limit intestinal absorption, whereas high lipophilicity can enhance the intestinal absorption of a drug. Other factors like permeability and formulation play significant roles in determining the overall bioavailability of a drug. Strategies such as the use of surfactants, lipids, permeation enhancers, micronization, salt formation, nanoparticles-drug delivery system, and solid dispersions are employed to overcome issues related to poor aqueous solubility and enhance the bioavailability of lipophilic drugs [41,42,43]. Thus, PVL compounds can be potentially used as oral administration drugs. However, further studies are needed to optimize the absorption capacity for future drug development.
It is noteworthy that all PVL compounds have low water solubility (< 0.4 mol/L), indicating poor water solubility related to lipophilicity. However, all compounds have high intestinal absorption in humans, indicating the percentage of drug absorption in the small intestine. Thus, further studies to optimize the absorption capacity are needed for future drug development.
The distribution property shows the distribution of drugs within different body compartments. All PVL drugs have a low blood–brain barrier (BBB) (< 0.1 log BB) and central nervous system (CNS) permeability (< − 3 log PS), indicating that they cannot penetrate the BBB. Metabolic property is another essential factor that affects a drug's pharmacokinetics, pharmacodynamics, and safety profile. Cytochrome P450 is a primary component in drug metabolism located in the liver and intestine and is either induced or inhibited by various substances [40]. Most of the compounds contained in PVL do not inhibit the cytochromes, except for CYP1A2 and CYP3A4 in several substances as shown in Table 3.
Toxicity prediction of phytochemical constituents of P. vulgaris LToxicity analysis is an essential parameter in determining the safety of compounds. To know the safety of each compound, we predicted the possibility of toxicity using the OSIRIS software, which used some parameters such as mutagenic, tumorigenic, irritant, and reproductive effects. Mutagenic and tumorigenic are parameters to predict the effect of the compound on becoming mutagenic and causing tumors. As a result, most PVL compounds are safe for toxicity analysis instead of quercetin, kaempferol, myricetin, 3,4-dihydroxybenzoic acid, and daidzin. These compounds were predicted to have medium-risk mutagenic and tumorigenic effects, except for daidzin, which has high-risk tumorigenic and reproductive effects. In addition, myricetin-3-glycoside, quercetin-3-glycoside, kaempferol-3-glycoside, catechin, and tannic acid have no risk for toxicity analysis, as shown in Fig. 1B. Nevertheless, wet lab studies are required to determine the optimal dosage, and a significant risk depends on the amount (16).
Membrane permeability prediction phytochemical constituents of P. vulgaris LThe ability of compounds to penetrate the membrane revealed that the compound with the lowest transfer energy along the translocation pathways identified by its calculated transfer energy profile is more permeable across the bilayer membrane [23]. To know the membrane penetration ability of each compound, we predicted using the PerMM web server. Figure 1C is a visualization of the conformational change of the PVL active compounds as they penetrated the cell membrane. Each molecule continuously adjusted its position to match the hydrophilic and hydrophobic characteristics of the plasma membrane. The analysis showed that plerixafor had the lowest energy across the membrane. Among the compounds contained in PVL, 3,4-dihydroxybenzoic acid has the lowest energy to cross the membrane, followed by kaempferol, daidzin, catechin, tannic acid, quercetin, myricetin, kaempferol-3-glucoside, quercetin-3-glucoside, and myricetin-3-glucoside, as shown in Fig. 1D.
Hydrophobic (non-polar) compounds can easily penetrate the lipid bilayer. Hydrophilic (polar) molecules, on the other hand, typically rely on transport proteins to cross the membrane because they cannot easily penetrate the hydrophobic interior of the bilayer [23]. Typically, the ligand for CXCR4 does not enter the membrane. Instead, it attaches to the extracellular region of the receptor, inducing a structural change that transmits a signal throughout the cell.
CXCR4 and CXCL12 binding mode interaction and plerixafor as inhibitorThe protein–protein interaction results were then analyzed for quality using Ramachandran plots via the PROCHECK web server. A good quality model was confirmed based on residues in the most favored regions (> 90%), as shown in Additional file 1: Fig. S1. The interaction appears to be similar to previous studies in that CXCL12 bound to the extracellular region of CXCR4 contains a significant negative potential, including critical negatively charged residues at residues Asp187, Asp97, Asp262, and Glu288 [10, 11].
As we mentioned in the introduction plerixafor is a CXCR4 inhibitor. The visualization interaction between plerixafor and CXCR4 shows that it can bind to the extracellular region of CXCR4 (Fig. 2B) at Asp187, Asp97, and Glu288 residue, but slightly far from Asp262 residue as shown in Figs. 2C. The mechanism by which SDF-1 binds to CXCR4 involves the interaction of the N-terminus domain of SDF-1 with the extracellular domain CXCR4, followed by further interactions that stabilize the complex and lead to signal transduction. Superimposed interaction shows that plerixafor has a binding mode similar to CXLC12. This binding mode curled up in the CXCR4 extracellular binding pocket of the CXCr4 [44] as shown in Fig. 2D. 2D interaction shows that plerixafor can bin to CXCR4 by forming hydrogen bond at Glu288, Tyr45, Asp97, Leu41, Ile185, Arg30, His281, Ala98, Ser285, Cys186, Trp102, Val112, Asp187, Arg188; hydrophobic bond at His113 and Trp94 as shown in Fig. 2E. As we mention before that Glu288, Asp187 and Asp97 is important residue in binding interaction CXCR4/CXCL12, indicating the reasonable of plerixafor as an established drug for CXCR4 antagonist. Moreover, Plerixafor is the only CXCR4 antagonist approved by the FDA and has been commercially distributed. Several studies have developed potential candidates for synthetic CXCR4 antagonists, such as POL6326 (Balixafortide), LY2510924, TN14003, and MSX-122, which exhibit good safety and tolerability profiles yet are still in preclinical and clinical studies phases [45,46,47]. So, we used plerixafor as the control ligand in this study.
Molecular docking before and after dynamics simulation of P. vulgaris L active constituent against CXCR4A molecular docking study is a research model used in drug discovery to determine the binding interaction between ligand and protein [48]. The binding affinity measures the strength of the interaction between two molecules [49]. Our molecular docking analysis identified that quercetin (− 6.6 kcal/mol), myricetin (− 6.6 kcal/mol), kaempferol (− 6.3 kcal/mol), catechin (-6.5 kcal/mol), and 3,4-dihydroxybenzoic acid (− 5.4 kcal/mol) bind to CXCR4 with the highest affinity compared to plerixafor (− 5.0 kcal/mol) as the control ligand. The remaining compounds with the lowest affinity compared to the control ligand are myricetin-3-glucoside (− 2.5 kcal/mol), quercetin-3-glucoside (− 0.6 kcal/mol), kaempferol-3-glucoside (− 2.5 kcal/mol), daidzin (− 2.8 kcal/mol), and tannic acid (− 4.2 kcal/mol), with lower affinity compared to plerixafor. The molecule with the lowest binding energy will have a constant temperature and pressure, called a stable molecule. The amino acid residues influenced the binding domain of the target protein and the type of chemical interactions in the binding region [50]. A lower energy value of binding affinity denotes a more stable and favorable binding relationship between the target protein and the ligand [49]. However, further experimental studies are necessary to confirm the actual protein–ligand interaction [51].
The interaction between catechin and CXCR4 formed a hydrogen bond at Glu32, Leu41, Tyr45, Val112, Tyr116, Arg183, Ile185, Ser285, and Glu288; a hydrophobic bond at Trp94, Ala98, and Asp97 and His113 (Fig. 5A). In addition, myricetin binds to CXCR4, forming a hydrogen bond with Arg30, Glu32, Phe93, Trp94, Asp97, Trp102, Val112, Tyr126, Ser285, and Arg188; a hydrophobic bond with His113, and an unfavorable donor bond with His281 (Fig. 5B), while, 3,4-dihydroxybenzoic acid can interact through binding interactions with Trp102, Val112, Tyr116, Asp97, Arg188, Hlu288 via hydrogen, Trp94 via hydrophobic, and His113 via pi-anion bonds (Fig. 5C). Kaempferol interactions at Arg30, Glu32, Asp97, Val112, Arg188, His281, and Ser285 as hydrogen bonds; Trp94 and Tyr116 as hydrophobic bonds; and His113 and Glu288 as pi-anion bonds (Fig. 5D). Kaempferol and plerixafor have the same binding interaction in Trp94 via a hydrophobic bond and in Glu288 via a pi-anion bond. Then, the quercetin-CXCR4 interaction formed a hydrogen bond at Leu42, Tyr45, Val112, Tyr116, His281, Asp187, Ser285, and Glu288; a hydrophobic bond at Trp94 and His113; and a pi-anion bond with Asp97 (Fig. 5E). All compounds have similar interactions compared to plerixafor. They can bind at least in Asp97 and Glu288, which have critical binding in the CXCL12/CXCR4 interaction.
Molecular dynamic simulations were performed for 20 ns to evaluate the structural behavior of the lead compounds within the substrate-binding active cavity of CXCR4. Plerixafor post-MD interaction forms a hydrogen bond at Arg30, Thr90, Phe93, Asp97, Trp102, Cys109, Val112, Arg188, Tyr190, Ile284, Gln200, Asp262, and Glu288; hydrophobic bond at Trp94, His113, Tyr116 and Cys186 as shown in Fig. 4. Cathecin post-MD interaction forms a hydrogen bond at Phe87, Leu91, Thr90, Phe93, Cys109, Cys186, His113, Ile185, Tyr255, Ile259, and Phe292; a hydrophobic bond at Trp94, Trp102, Tyr116, and Val112 (Fig. 5F). While myricetin, post-MD simulation, forms a hydrogen bond with Thr90, Phe93, Asp97, His113, Ala175, Asn176, Cys186, Asp187, Arg188, Glu288; and a hydrophobic bond with Trp94, Trp102, Val112, and Tyr116 as shown in Fig. 5G. 3,4-dihydroxybenzoic acid post-MD interaction is formed via a hydrogen bond with Thr90, Phe93, Asp97, Trp102, His113, Tyr116, Cys186; and a hydrophobic bond with Trp94 and Val112 (Fig. 5H). In addition, based on MD simulations, kaempferol-CXCR4 interacts by forming a hydrogen bond with Phe13, Glu15, Ser16, Trp57, Leu55, and Val18; and a hydrophobic bond with Phe14 (Fig. 5I), while quercetin post-MD simulation formed a hydrogen bond at Ser28, Leu41, Tyr45, Val112, Tyr116, and Arg188; a hydrophobic bond at Trp94 and Ala98; and a pi-anion bond with Asp97 and His113 (Fig. 5J). Post-MD interaction analysis showed that quercetin, myricetin, and 3,4-dihydroxybenzoic acid still can bind to critical amino acid residue at Asp97, Asp187, and Glu288 as shown in Table 4 and Fig. 5. The stability of the CXCR4 protein refers to the collective pressures determining whether the protein will maintain its folded shape or adopt non-native aggregating configurations. Comparing the structure of protein–ligand complexes at different levels of stimulation up to 20 ns provides valuable structural insights that help to understand the potential changes in ligand position that occur as a result.
Fig. 4
2D Visualization post-MD (Molecular dynamic simulations) of Plerixafor against CXCR4
Fig. 5
Amino acid residues result from the interaction between ligands and CXCR4. Panels A to E show interaction before molecular dynamics, while panels F to J show residues after molecular dynamics simulations
Table 4 Molecular interaction active compound of P. vulgaris L. with CXCR4The protein–ligand interactions play a vital role in structural biology by providing insight into the mechanisms of these interactions at the molecular level. The similarity of the binding to the control indicates the same function. A similar interaction active constituent of PVL can be seen in Table 4. Identification of molecular interactions and binding orientations on the docked protein–ligand complex revealed that PVL compounds form non-covalent interactions with all target proteins through hydrophobic, pi, and hydrogen bonds. These interactions lead to the development of the protein–ligand complex and trigger the initiation of an activity response, including enhancement and inhibition of the target protein [22, 48]. Together with another additional factor, such as binding affinity (binding affinity analysis), the similarity of those interaction types between a natural compound and the control ligand (interaction analysis) could indicate the accuracy of the docking method and support the potential of a ligand as a drug candidate [52, 53]. The critical molecular interaction, hydrophobic, pi, and hydrogen bonds can help to stabilize the ligand within the protein's active site, leading to a stronger binding affinity and potentially inhibitory activity [53, 54]. The similarity interaction between the PVL compound and the control ligand (Plerixafor) suggests the potential inhibitory activity as a CXCR4 antagonist candidate. Since molecular docking solely is insufficient to conclude the promising activity, further methods such as molecular dynamic simulation will provide information about the stability and dynamics of the protein–ligand complex by optimizing the structures of the final complexes from docking, calculating detailed interaction energies, and providing information about the ligand binding mechanism [55], as we conducted in this study.
The molecular dynamics simulation2D plots were generated to illustrate the varying behavior of the docked complex at different time intervals during the MD simulation runs. The plots play a critical role in the statistical analysis of the MD simulations. They provide valuable insight into the stability and flexibility of the residues at different time points during the simulations [56]. The RMSD results show a stable interaction between plerixafor and CXCR4. The value was approximately 0.3 nm from 0 to 20 ns. The active constituent of PVL, catechin RMSD, shows stability from 2 to 11 ns with an average RMSD value of 1.1 nm, then unstable fluctuation up to 20 ns. Myricetin shows instability from 0 to 20 ns with an average RMSD value of 1 nm, indicating that the interaction of myricetin with CXCR4 is unstable. 3,4-dihydroxybenzoic acid fluctuates from 0 to 6 ns and is stable from 6 to 20 with an RMSD score of 0.7 nm. Kaemferol was stable from 3 to 12 ns with an RMSD score of 0.4 nm, then fluctuated to 16 ns and stabilized at 20 ns. Interestingly, quercetin has a similar stability interaction with an average RMSD score of 0.3 nm from 0 to 20 ns, indicating that quercetin has a similar stability interaction compared to plerixafor, as shown in Fig. 6A.
Fig. 6
The molecular dynamic of PVL against CXCR4. A Root Mean Square (RMSD), B Root Mean Square Fluctuation (RMSF), C Number of Hydrogen bond
The PVL compound causes significant fluctuations in amino acid residues in certain regions of the protein, as shown by the RMSF curve in Fig. 6B. Analysis of residue-specific root mean square fluctuation (RMSF) provides insight into how ligand binding affects the structural flexibility of a protein at the single amino acid level [57]. A higher RMSF value indicates greater flexibility of the complex. Leu68, Asp97, Leu146, and Thr178 have the largest backbone variation of 5 nm. The significant variations in amino acid residues suggest the fluctuating interactions between the compound and these residues, possibly involving hydrogen bonding, ionic interactions, or van der Waals forces. The PVL compounds appear to have a remarkable effect on the structure and dynamics of the protein, potentially serving as a modulator or regulator of important protein functions.
To explain the conformational stability of the interaction, we examine the total number of intermolecular hydrogen bonds in the ligand–protein complexes. Hydrogen bonds play an important role in the stability of the protein structure [55]. Interestingly, the number of hydrogen bonds in catechin, hydroxybenzoic acid, myricetin, and quercetin compounds has the same value as plerixafor, with an average of 380. but the number of hydrogen bonds in kaempferol compounds has a lower average of 320, as shown in Fig. 6C.
Future potential active constituent of PVL as CXCR4 inhibitorPVL is rich in a variety of phytochemicals, including flavonoids, phenolic acids, and other antioxidants [17]. Some of these compounds have shown biological activity in various contexts, although their specific interaction with CXCR4 would require targeted research.
The CXCR4/CXCL12 pathway has been proposed as a target for stem cell mobilization-based therapy in various rheumatic diseases. Several studies have reported that upregulation of CXCR4 and CXCL12 is associated with joint erosion, synovial inflammation, synovial hyperplasia, and synovial angiogenesis [58]. Mesenchymal stem cells have been shown to stimulate chondrocyte regeneration and differentiation into cartilage [59]. Osteoarthritis, stem cell-based treatment has shown promising clinical effects, including improved joint function, pain threshold, and quality of life [60]. In comparison, stem cell-based treatment can modify and restore the balance of inflammatory T cells in rheumatoid arthritis [61].
CXCL12/CXCR4 axis also has a potential therapeutic target for several inflammatory diseases, not only by affecting cell migration but also by altering the immune response. Only one antagonist targeting the CXCR4 ligand binding region, plerixafor, has shown therapeutic relevance (1). In addition, the role of chemokines in immune modulation in autoimmune diseases remains to be explored. Chronic diseases, especially autoimmune diseases such as rheumatoid arthritis (RA), inflammatory bowel disease (IBD), and systemic lupus erythematosus (SLE), are associated with an abnormal inflammatory response because the immune system recognizes the protein as an antigen and attacks itself [62,63,64].
This study shows that several candidate active constituents of PVL can potentially become CXCR4 inhibitors. Computational tools and molecular modeling can predict the interaction between potential phytochemicals in PVL against the CXCR4 receptor. Such in silico studies can be a cost-effective first step before experimental studies, but this finding needs to be clarified with more precise and advanced studies [65]. Any potential CXCR4 inhibitors identified would need to undergo rigorous preclinical and clinical testing to establish their safety and efficacy.
The efficacy of natural product derived-CXCR4 inhibitor flavonoids, isoflavones, bioketones, and isoprenoidyl has been observed through CXCR12/CXCR4 axis. Promising inhibitory activity of flavonoid compounds such as Quercetin [66, 67] and myricetin has been reported in downregulating CXCL12 and CXCR4 expression in prostate cancer [68]. However, further safety assessment is necessary to validate those efficacious activities of the novel CXCR4 antagonist. Research in CXCR4 antagonist development from natural products is escalating despite the compound's pharmacokinetic, pharmacodynamic, and toxicity testing not being comprehensively studied, which is insufficient to replace the established agent Plerixafor.
We understand that there are limitations to this study. In our study, we only predicted the potential of the active component of PVL against CXCR4 using computational study. Therefore, we cannot confirm the effect of PVL on CXCR4 in experimental studies using cells or animals. At least, this research can support and serve as a basis for further research in vitro and in vivo. Further research is needed to clarify our predictive findings through experimental studies, such as examination of binding interaction and visualization using X-rays or cryo-EM. Experimental toxicity studies are also needed to confirm PVL as a candidate for CXCR4 inhibitor. It is important to note that while natural products are a promising source for drug discovery, the path from identifying a potential lead compound to developing a clinically approved drug is long, complex, and challenging. Any findings would need to be substantiated through rigorous scientific research and clinical trials. At this time, any potential CXCR4 inhibitory compounds in PVL remain speculative and would require significant research to validate.
留言 (0)