Test substances
HemoHIM G (Lot No. 2100001) was prepared by concentrating extract under reduced pressure to achieve an optimal concentration. No additional additives were introduced during this process. The concentrated extract was then transformed into a powdered form through lyophilization. HemoHIM G extract is checked and standardized for its consistency with chlorogenic acid (more than 80% of 0.216 mg/g) derived from Angelica gigas and Ligusticum chuanxiong, and paeoniflorin (more than 80% of 2.869 mg/g) derived from Paeonia lactiflora, as marker substances, manufactured by Kolmar BNH Co. Ltd. (Sejong, Korea). The sample for safety test contains chlorogenic acid 0.216 mg/g, and paeoniflorin 2.869 mg/g. In addition to the marker substances, ferulic acid and (Z)-ligustilide from Angelica gigas and Ligusticum chuanxiong, senkyunolide derivatives such as senkyunolide A and H from Ligusticum chuanxiong, and paeoniflorin, albiflorin, and gallic acid from Paeonia lactiflora were confirmed to be contained in HemoHIM G. The formulation was prepared immediately before administration, on the same day it was intended to be administered.
Animals and husbandry
All rats (Crl:CD(SD), 6 weeks old) for this studies were obtained from Orientbio Inc. (Seongnam, Korea). Environmental conditions in the animal room were maintained as follows: temperature = 19–25 °C, relative humidity = 30–70%, air exchange rate 10–15 changes/h, and light/dark cycle = 12 h/12 h. Variations in these conditions had no effect on the study outcomes.
This study was conducted in accordance with the following Good Laboratory Practice Regulations: “Good Laboratory Practice Regulation for Nonclinical Laboratory Studies”, Notification No. 2018–93, Ministry of Food and Drug Safety, Republic of Korea (Nov. 21, 2018); “OECD Principles of Good Laboratory Practice”, Organization for Economic Co-operation and Development (OECD), ENV/MC/CHEM(98)17 (as revised in 1997).
This study was conducted at Biotoxtech Co., Ltd. (Cheongju, Korea), which received full accreditation from the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC International) in 2010. This study was reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of Biotoxtech Co., Ltd. based on Animal Protection Act of Republic of Korea (Enactment May 31, 1991, No. 4379, Revision Feb. 11, 2020, No.16977) (Approval No.: 220298).
Acute oral toxicity
An acute oral toxicity study was conducted in accordance with Organization for Economic Cooperation and Development (OECD) Guideline 423 following the application of good laboratory practice (GLP) [16]. The acute toxicity of HemoHIM G was assessed in male and female SD rats via oral gavage, with the test substance dissolved in water for injection and administered at the dose limit of the preliminary study (5000 mg/10 mL/kg of body weight). All animals were fasted overnight (16 h) (water adlibitum with no feed) before administrating the test substance. All animals were observed for mortality, morbidity, and signs of toxicity (clinical signs) at 30 min, 1, 2, 4, and 6 h after dosing on day 0 and once daily thereafter for 14 days. Body weights were recorded prior to dosing on days 0, 2, 4, 8, and 15. At the end of the 14-day observation period, necropsy and gross pathological examinations were performed.
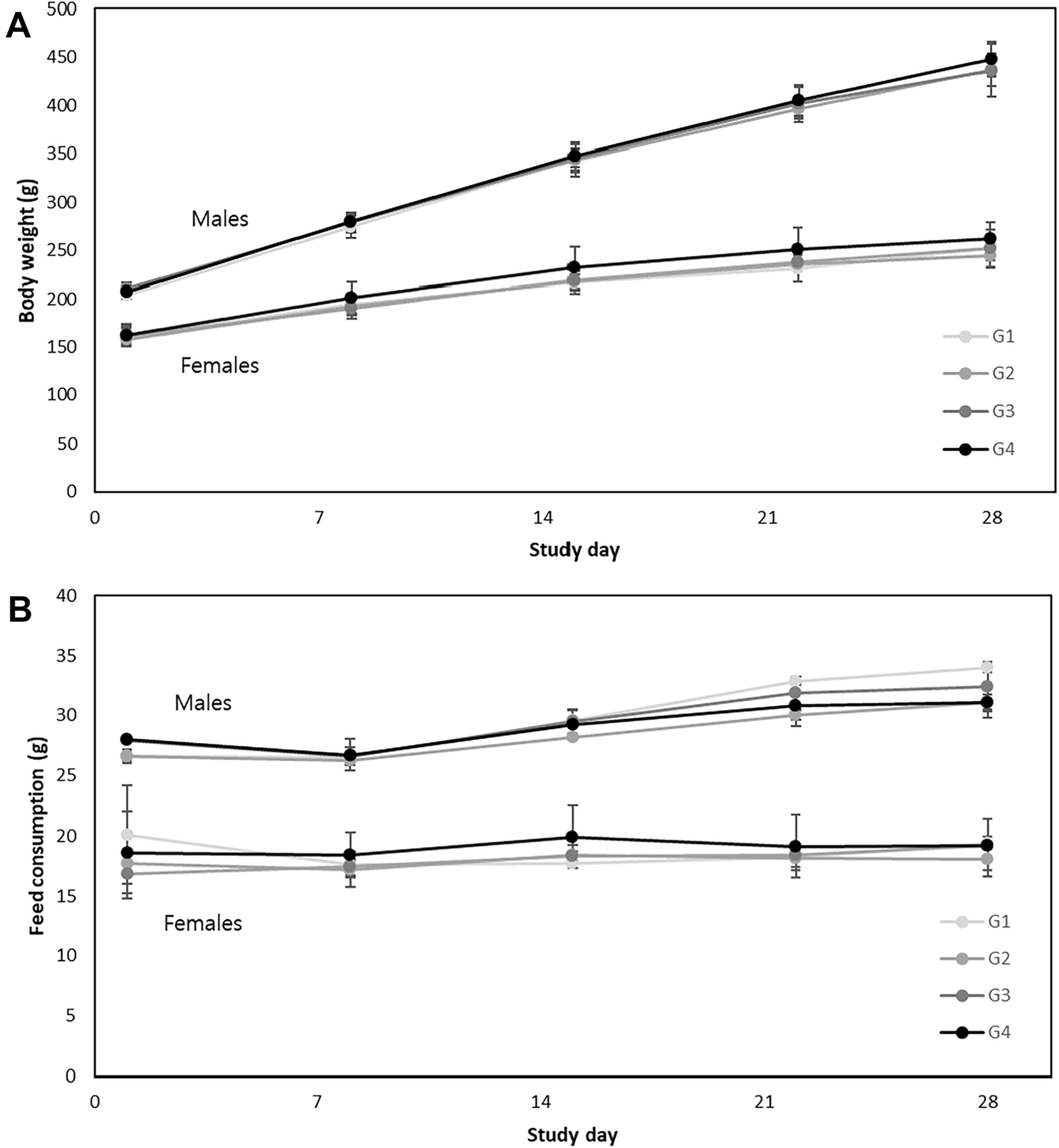
28-Day repeated dose oral toxicity
A repeated-dose oral toxicity study was conducted in accordance with the OECD Guideline 407 following the application of GLP [17]. The doses were administered orally to SD rats for 28 consecutive days to assess of any toxic effects. The test substances were weighed, suspended in water, and administered to rats through the oral (gavage) route using a disposable syringe with a rat intubation cannula at graduated dose levels of 1250 mg/kg/day for low dose (G2), 2500 mg/kg/day for mid-dose (G3), and 5000 mg/kg/day for high dose (G4). The rats in the control group (G1) received water alone. The administered dose volume was 10 mL/kg/day. Each group consisted of five rats of each sex. Vehicle or test formulations were administered to each rat group once daily for 28 consecutive days. The animals were observed twice daily for mortality/morbidity and once daily for cage-side clinical signs. Detailed clinical examinations were performed once prior to the initiation of treatment and thereafter at weekly intervals and the end of the treatment and recovery periods. The rats were observed once per week for changes in body weight and feed consumption. Hematological and clinical chemistry investigations were performed at the end of the treatment and recovery periods.
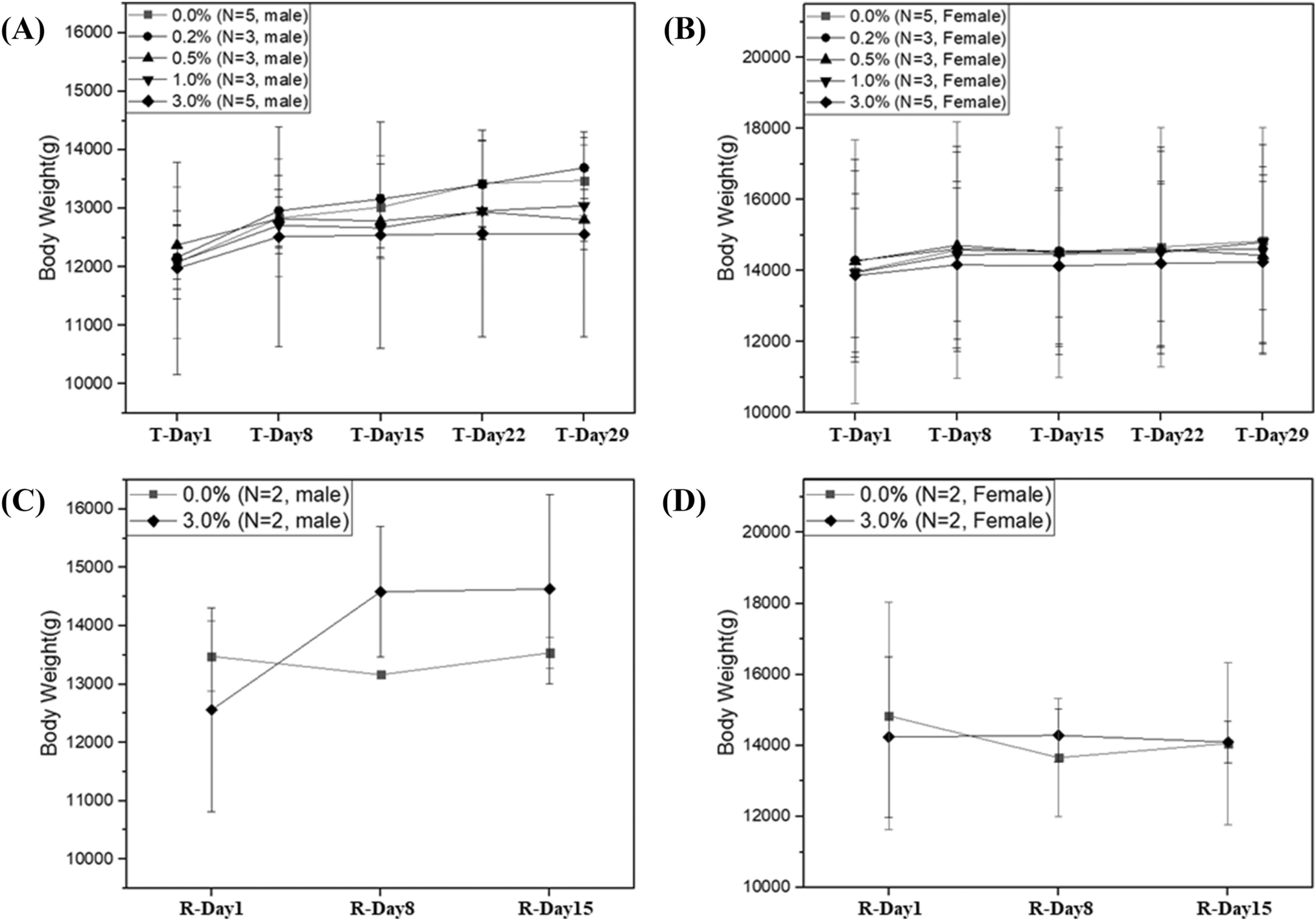
13-Week repeated dose oral toxicity
A repeated-dose oral toxicity study was conducted in accordance with OECD Guideline 408 following the application of GLP [18]. The doses were administered orally to SD rats for 13 consecutive weeks, followed by a 28-day recovery period to assess the reversibility of any toxic effects. The test substance was weighed, suspended in water, and administered to rats through the oral (gavage) route using a disposable syringe with a rat intubation cannula at graduated dose levels of 1250 mg/kg/day for low dose (G2), 2500 mg/kg/day for mid-dose (G3), 5000 mg/kg/day for high dose (G4), and high dose recovery groups (G4R). The rats in the control group (G1) and control recovery group (G1R) received water alone. The administered dose volume was 10 mL/kg/day. Each group consisted of 10 rats of each sex. Vehicle or test formulations were administered to each rat group once daily for 13 consecutive weeks. The animals were observed twice daily for mortality/morbidity and once daily for cage-side clinical signs. A detailed clinical examination was performed once prior to the initiation of treatment, thereafter at weekly intervals, and the end of the treatment. The rats were observed once per week for changes in body weight and feed consumption. Hematological and clinical chemistry investigations were performed at the end of treatment.
Bacterial reverse mutation assay
An in vitro bacterial reverse mutation assay was conducted in accordance with OECD Guideline 471 following the application of GLP [19]. In preliminary cytotoxicity assay, TA1537, TA1535, TA100, TA98 of Salmonella typhimurium strain were treated with the test substance at the concentrations of 313, 625, 1250, 2500, and 5000 μg/plate both in the presence (S9 mix) and absence of metabolic activation system. Vehicle and positive controls were maintained concurrently with the treatment groups. Based on the results observed in the preliminary cytotoxicity assay, 5000.0 μg/plate was selected as the highest concentration for mutagenicity assay. Mutagenicity assays were performed using the TA1537, TA1535, TA98, and TA100 strains of S. typhimurium and the WP2uvrA strain of E. coli. The bacterial strains were treated with the test substance at 313, 625, 1250, 2500, and 5000 μg/plate in the presence (S9 mix) and absence of a metabolic activation system.
In vitro mammalian chromosomal aberration assay
An in vitro mammalian chromosomal aberration assay was conducted in accordance with OECD Guideline 473 following the application of GLP [20]. Based on the preliminary cytotoxicity assay results, chromosome aberration assay was conducted using three different concentrations of test substance i.e., 78, 156, 313, and 625 μg/mL in the presence and absence of a metabolic activation system. Benzo[a]pyrene (with the metabolic activation system S9) and mitomycin C (without the metabolic activation system S9) were used as clastogenic positive controls. Chinese hamster lung cell (CHL/IU cell) were cultured using Eagle’s minimum essential medium supplemented with 10% FBS, 1% penicillin–Streptomycin, in CO2 incubator, at 37 ± 1 °C and 5 ± 0.5% CO2. These cultures were exposed to different concentrations of test substances for short-term (6 h) and continuous (24 h) exposure. In short-term exposure, after 6 h of treatment, culture media with test substance was replaced with fresh medium and further incubated for 18 h at 37 ± 1 °C and 5 ± 0.5% CO2. For continuous exposure, cultured cells were treated with different concentrations of test compounds for 24 h. After 24 h, cultures from the short-term and continuous exposure groups were harvested and processed for slide preparation. The slides were stained with Giemsa stain (3%, v/v). Slides were observed in the order of short-term and continuous treatments. Chromosomal aberrations were classified as structural aberrations, numerical aberrations, etc.
Mammalian bone marrow erythrocyte micronucleus assay
An in vivo mammalian bone marrow erythrocyte micronucleus assay was conducted in accordance with OECD Guideline 474 following the application of GLP [21]. Micronucleus tests were conducted at doses of 1250, 2500, and 5000 mg/kg. The dose levels for the micronucleus test were selected based on a dose-range finding study. For the micronucleus test, HemoHIM G was orally administered to SD rats at a dose volume of 10 mL/kg for 2 days, with an interval of approximately 24 h. Animals in the positive control group received a single dose of cyclophosphamide orally at 20 mg/kg/day before bone marrow collection. Approximately 24 h after dosing, all animals were euthanized and both femur bones were collected from each animal. The bone marrow was collected using phosphate buffered saline (PBS). After collection, all samples were centrifuged, and the supernatant was discarded, leaving a small amount of PBS cell pellet. Smears were prepared on slides using the cell pellet. The slides were air-dried, fixed in 10% neutral formalin, and stained with 0.05% acridine orange.
Statistical method
Statistical analysis was performed on the data of body weight, feed consumption, urine volume, hematology, clinical chemistry and organ weights using SAS program (version 9.4, SAS Institute Inc., USA). For the main groups and dosing period, the data was analyzed by Bartlett’s test for homogeneity of variance (significance level: 0.05). One-way analysis of variance (ANOVA) was employed on homogeneous data; then, if significant (significance level: 0.05), Dunnett’s test was applied for multiple comparisons (significance levels: 0.05 and 0.01, two-tailed). Kruskal–Wallis test was employed on heterogeneous data; then, if significant (significance level: 0.05), DSCF (Dwass–Steel–Critchlow–Fligner) was applied for multiple comparisons (significance levels: 0.05 and 0.01, two-tailed). For the recovery groups, the data was analyzed utilizing Folded F-test for homogeneity of variance (significance level: 0.05). Student’s t-test was employed on homogeneous data or Aspin-Welch t-test was employed on heterogeneous data for confirming significance (significance levels: 0.05 and 0.01, two-tailed).









留言 (0)