
記住我



This study was conducted in compliance with the ethical principles originating in or derived from the Declaration of Helsinki and in compliance with all International Conference on Harmonization Good Clinical Practice Guidelines. In addition, all local regulatory requirements were followed, particularly those affording greater protection to the safety of trial participants.
Male or female participants were eligible for enrollment if they were ≥ 18 years of age; had histologically confirmed diagnosis of locally advanced, unresectable or metastatic cutaneous melanoma American Joint Committee on Cancer (AJCC) stage IIIB, IIIC, or IV, or other BRAF V600-mutant advanced solid tumors, with evidence of measurable or non-measurable lesions as detected by radiological or photographic methods according to guidelines based on Response Evaluation Criteria in Solid Tumors 1.1; had Eastern Cooperative Oncology Group (ECOG) performance status (PS) of 0 or 1; and had adequate bone marrow, hepatic, and renal function. Use of contraception was necessary for both males and females of childbearing potential. The type of contraception allowed in this study was determined to be effective and acceptable per Clinical Trial Facilitation Group guidance.
Key exclusion criteria included symptomatic brain metastasis, history of reaction to any of the study medications, symptomatic or untreated leptomeningeal disease, history or current evidence of or current risk factors for retinal vein occlusion (e.g., uncontrolled glaucoma or ocular hypertension, history of hyperviscosity or hypercoagulability syndromes), clinically significant cardiac disease, impaired hepatic function as defined by Child-Pugh class B or C, impaired gastrointestinal function or disease which might have significantly altered the absorption of study drugs (e.g., ulcerative diseases, uncontrolled nausea, vomiting, diarrhea, malabsorption syndrome, and small bowel resection), known hypercoagulability risks other than malignancy (e.g., factor V Leiden syndrome), thromboembolic event except catheter-related venous thrombosis ≤ 12 weeks prior to starting study treatment, discontinuation of prior BRAF and/or MEK inhibitor treatment owing to left ventricular dysfunction, pneumonitis/interstitial lung disease, or retinal vein occlusion.
Participants were also excluded if they had used any herbal medications/supplements or any medications or foods that are moderate or strong inhibitors or inducers of CYP3A4/5, or consumed grapefruit, pomegranates, star fruits, Seville oranges, or products containing the juice, or any substrates, inhibitors, or inducers of CYP2B6, or any substrates or inhibitors of BCRP, OATP1B1, or OATP1B3 within 2 weeks prior to the start of encorafenib/binimetinib treatment on day 1 and through DDI phase (day 28).
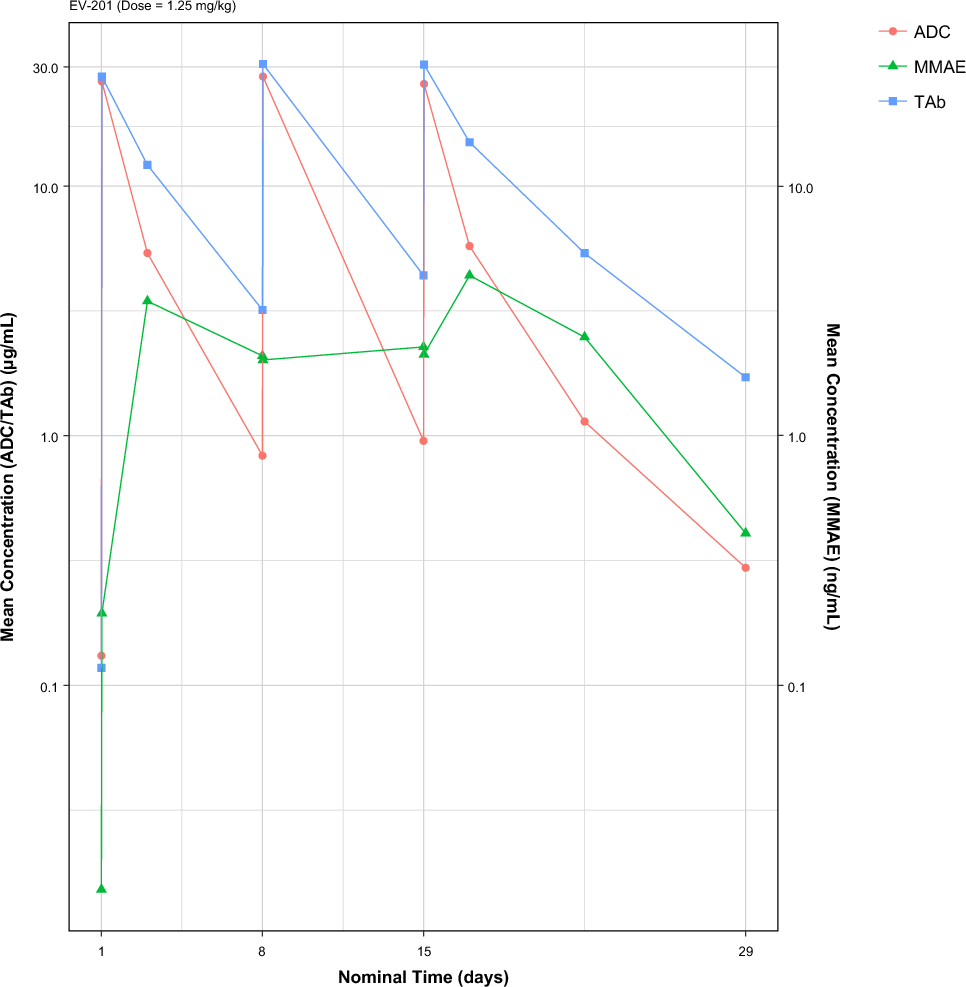
2.2 Phase 1 Study DesignThe phase 1 study design is illustrated in Fig. 1. Based on estimates of the within-subject variability for rosuvastatin exposure [18], a total of approximately ten participants were needed to meet the primary study objectives. Participants received a single oral dose of rosuvastatin 10 mg and immediate-release bupropion 75 mg on days − 7, 1, and 14. Encorafenib 450 mg once daily and binimetinib 45 mg twice daily were administered starting on day 1. Blood samples for measurement of plasma concentrations of encorafenib (and its metabolite, LHY746), binimetinib (and its metabolite, AR00426032), rosuvastatin, and bupropion (and its metabolite, hydroxybupropion) were collected at 0, 1, 2, 3, 4, 6, and 8 h post dose on days − 7, 1, and 14. The study drugs were administered in the fasted state, and participants were instructed to refrain from eating for 1 h following encorafenib and binimetinib intake.
Fig. 1
Phase 1 study design. PK pharmacokinetics
2.3 Plasma Sample Analysis and PK Parameter CalculationHuman PK plasma samples were analyzed for quantitation of all PK analytes at PPD (Middleton, WI, USA) using validated, sensitive, and specific high-performance liquid chromatography-mass spectrometry/mass spectrometry (HPLC-MS/MS) methods in compliance with laboratory standard operating procedures.
CP-I concentrations were measured retrospectively from remaining PK samples (0–6 h and sparse PK) collected from patients with colorectal cancer who received encorafenib and were analyzed using a validated HPLC-MS/MS. Further details on the study design are reported separately.
PK parameters including, but not limited to, Cmax and AUClast, were calculated for each participant using noncompartmental analysis methods using the WinNonlin software package (Phoenix WinNonlin Professional, version 8.0; Pharsight Corporation, Mountain View, CA, USA) for PK analytes, including rosuvastatin, bupropion, hydroxybupropion, encorafenib and its metabolite LHY746, and binimetinib and its metabolite AR00426032.
2.4 GenotypingEvaluation of the effect of polymorphisms on the exposure of rosuvastatin or bupropion was an exploratory objective in the study. Polymorphisms for OATP1B1 (SLCO1B1 rs2306283, rs4149056), BCRP (ABCG2 rs2231142, rs72552713), and CYP2B6 (*4,*5,*8,*9,*18,*22,*28) were analyzed on a QuantStudio 12K Flex Real-Time PCR System (Thermo Fisher Scientific, Waltham, MA, USA).
2.5 SafetySafety was assessed during the DDI phase (day − 7, 1, 14, and 28) and in the post DDI phase every 3–4 weeks until discontinuation of the study drug. Safety monitoring included serious adverse events (SAEs), laboratory profiles (hematology, biochemistry, coagulation, cardiac/muscle enzymes, urinalysis), physical examination (including vital signs, ophthalmic and dermatological examinations), ECOG PS assessment, cardiac profiles (electrocardiogram and multigated acquisition scan or echocardiogram), and concomitant medications or therapies. AEs were classified according to the Medical Dictionary for Regulatory Activities (http://www.meddra.org) classification system, version 22.1, and graded according to the Common Terminology Criteria for Adverse Events, version 4.03.
2.6 Statistical AnalysisFor the determination of sample size, the coefficient of variation of the ratio between two rosuvastatin AUClast values for the same participant for rosuvastatin is approximately 47% [18]. Assuming a two-sided significance level of 0.05 and a power of 0.8, approximately ten participants would need to be evaluated to detect a difference of 50% in mean AUClast. Assuming an intrasubject variation of 23% for bupropion AUC0-8, [20] there is an 80% probability with ten participants that a treatment difference will be detected if the true effect size is 33%.
All participants who received at least one dose of any study drug were included in the safety set population. The evaluable PK set included all participants with sufficient concentration data to calculate at least one PK parameter for a probe drug on days − 7, 1, and 14 (days 1 and 14 for binimetinib and encorafenib). Participants who discontinued, missed three or more consecutive doses of encorafenib prior to completion of the last PK sampling on day 14, or required a dose reduction of encorafenib prior to completion of the last PK sampling on day 14 were excluded from the evaluable PK set. In addition, participants who missed any dose of study drugs on any of the PK days or who vomited within 4 h after dosing on any of the PK days were excluded from the evaluable PK set.
An analysis of variance was performed on the natural log transformed Cmax and AUClast of rosuvastatin, bupropion, and hydroxybupropion. The least squares means geometric mean ratio (GMR) and associated 90% confidence interval (CI) for each PK parameter were calculated using the exponentiation of the difference between treatment least squares means from the analyses on the natural log transformed parameters and expressed as a percentage of day 1 relative to day − 7 and day 14 relative to day − 7.
2.7 Analysis of CP-I in Phase 3 Study ParticipantsCP-I concentrations were measured retrospectively from remaining PK samples (0–6-h profiles and sparse PK) collected from participants with colorectal cancer who received encorafenib in a separate phase 3 study and were analyzed using a validated HPLC-MS/MS. Further details on the study design are reported separately [14, 19]. Two approaches were used to evaluate the potential changes in CP-I concentrations: (1) to determine maximum fold change in the 0–6-h profiles in the safety lead in (SLI) phase participants in the phase 3 clinical study, and (2) to incorporate all of the CP-I patient data from this phase 3 study and use population compartmental modeling for a pooled analysis.
Nonlinear mixed effects models were developed in NONMEM (version 7.5.0) to fit individual participant encorafenib PK and pharmacodynamic (CP-I) profiles. Data formatting, postprocessing, and simulation-based sensitivity analysis were conducted in R Software (v3.6.1). Various PK/PD models were evaluated for their suitability to describe the CP-I concentration–time profiles, including those previously described [15, 17].
Model parameters were then estimated by utilizing encorafenib concentrations predicted on the basis of a previously described population PK model (Pfizer internal data). Encorafenib concentrations were converted to the unbound concentration (nM) on the basis of the molecular weight of encorafenib (540 Da) and the free fraction of drug (0.14) prior to estimation of CP-I model parameters. Final model pharmacodynamic parameters were used for a sensitivity analysis to evaluate the effect of varying the encorafenib in vivo inhibition constant (Ki) against OATP1B1 (range 1–10000 nM).
留言 (0)