
記住我
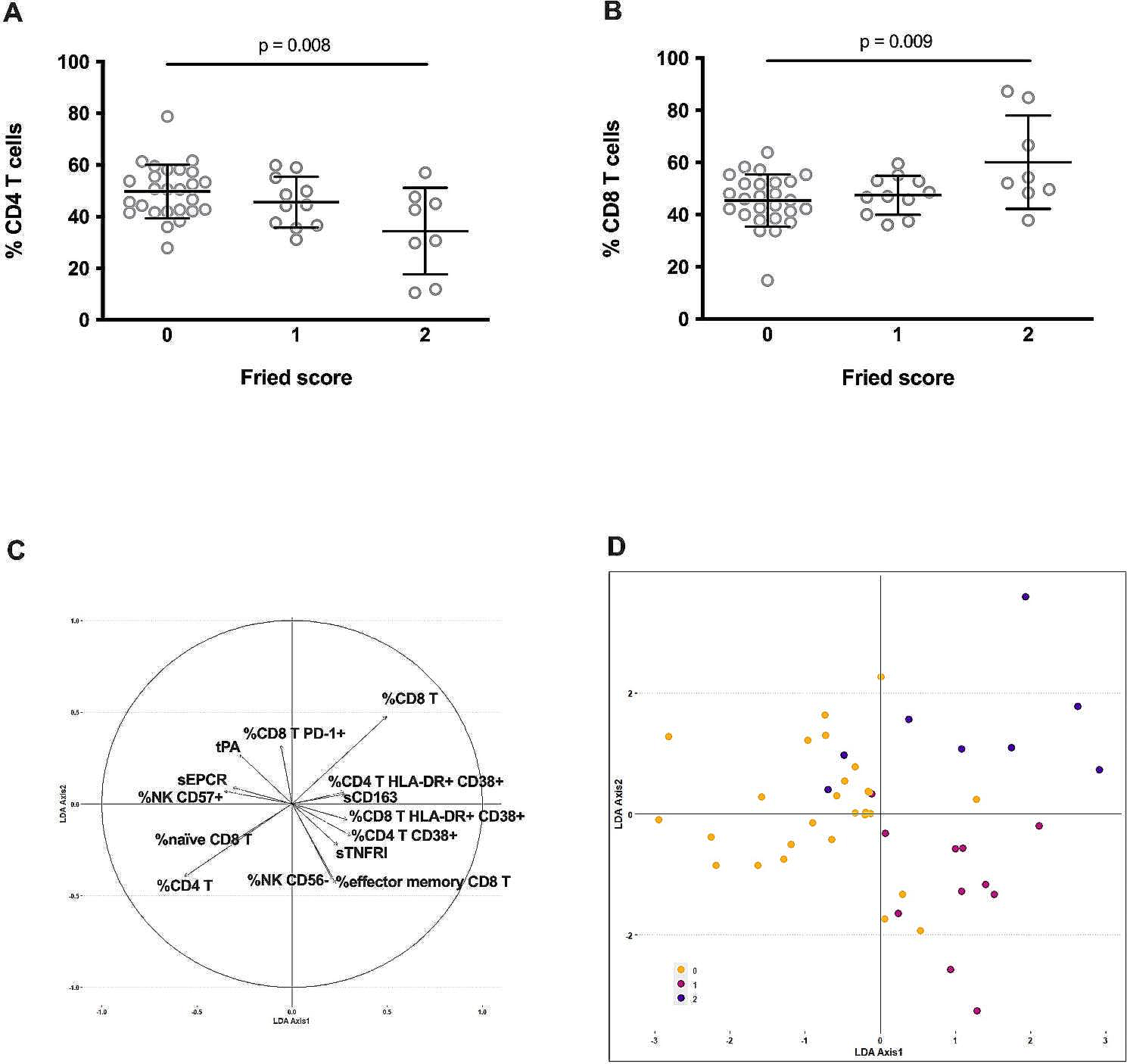
Much epidemiological evidence suggests that inflammation mediated by inflammatory molecules, including the NLRP3 inflammasome, is a powerful risk factor for cardiovascular disease.
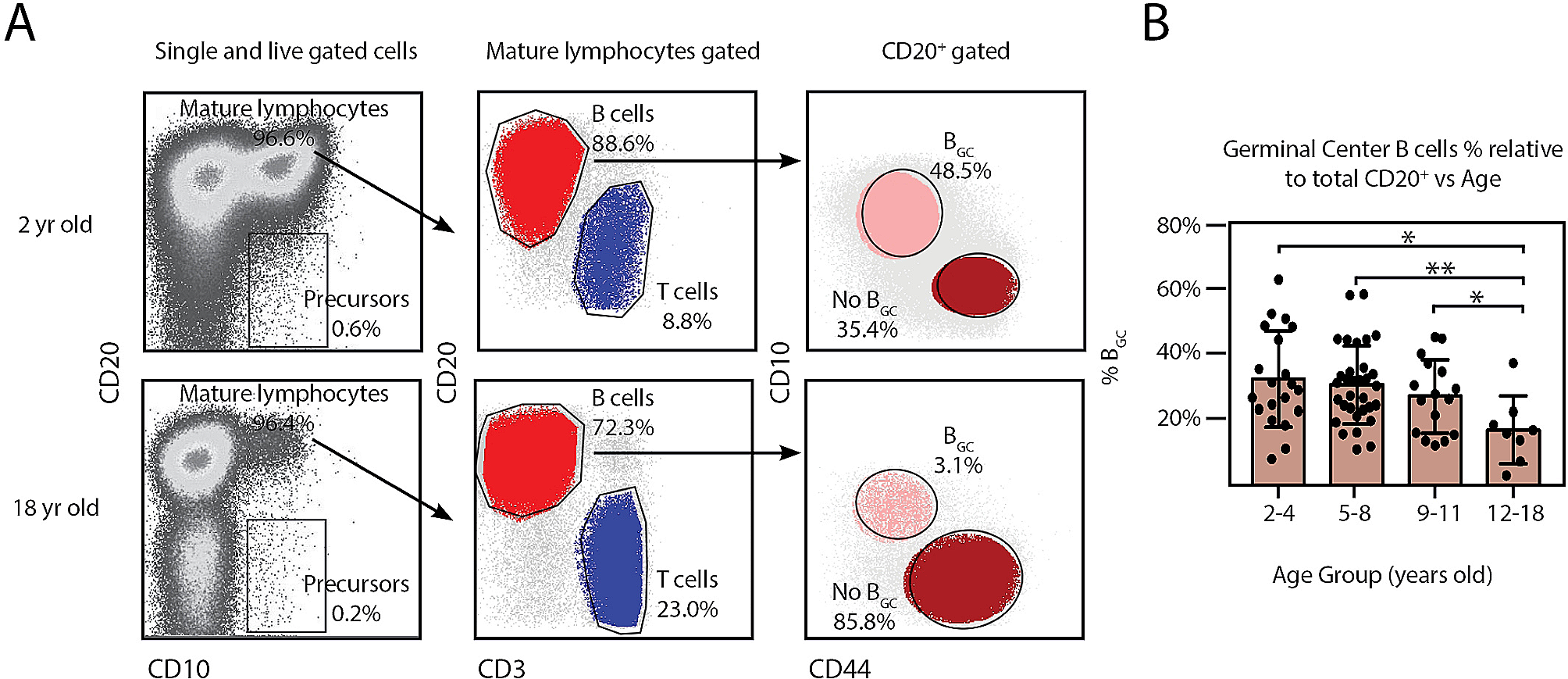
Atherosclerosis is an important manifestation of cardiovascular aging, as cells having markers of aging can be found in advanced atherosclerotic plaques. There is increasing evidence that inflammation is a crucial factor leading to atherosclerosis. The process of atherosclerosis is influenced by endogenous molecules, such as cholesterol crystals present in damaged vascular endothelial cells and other substances derived from damaged or necrotic tissue. These endogenous danger signals activate the NLRP3 inflammasome in macrophages, ultimately leading to the rupture of fragile atherosclerotic plaques, leading to thrombosis (Fig. 3) [35]. In addition, animal experiments in mice have found that the role of the NLRP3 inflammasome in atherosclerosis is due to its effector cytokine IL-1β. After feeding low density lipoprotein receptor knockout model mice with a high-fat diet for a period, it was found that their levels of IL-1β and the incidence of atherosclerosis were significantly higher than those of normal mice (Fig. 3). Loss of the low-density lipoprotein receptor results in the inability of cells to take up cholesterol in the plasma, thereby increasing plasma cholesterol concentrations. This suggests that the NLRP3 inflammasome can be activated by cholesterol in plasma [29]. A study by Marco Orecchioni et al. found that mouse plasma containing octanal activates the olfactory receptor Olfr2 expressed by vascular macrophages, driving atherogenesis through NLRP3-dependent IL-1 production (Fig. 3) [36].
Fig. 3
Activation of NLRP3 inflammasome in cardiovascular disease. AMPK, AMP-activated protein kinase; ASC, apoptosis-associated speck-like protein; IL-1, interleukin-1; IL-1β, interleukin-1β; LDL, low density lipoprotein; Olfr2, olfactory receptor 2
Cardiac ischemia–reperfusion injury is also associated with the occurrence of inflammatory responses (Fig. 3) [37]. In an experiment with metformin, the researchers found that AMPK, a protein kinase, can reduce myocardial ischemia–reperfusion injury by inhibiting the expression of the NLRP3 inflammasome and ASC [38]. Likewise, Meisoindigo is demonstrated to suppress NLRP3 inflammasome production, exhibiting a pivotal role in diminishing ischemia–reperfusion injury [39]. Collectively, these findings support the involvement of the NLRP3 inflammasome in promoting ischemia–reperfusion injury.
As an additional manifestation of aging, chronic heart failure in humans is intricately linked to inflammation, a relationship substantiated through animal model experiments. In a mouse model with Tet2 mutation [40], the NLRP3 inflammasome inhibitor MCC950 was administered, revealing that MCC950 prevented heart failure development (Fig. 3). Moreover, it eliminated disparities in cardiac parameters between Tet2-deficient and wild-type mice, ultimately reversing cardiac fibrosis and hypertrophy [40]. In another set of mouse experiments, OLT1177, an inhibitor of the NLRP3 inflammasome, partially restored cardiac function that was compromised in heart failure (Fig. 3) [41]. Despite these promising findings associating heart failure with inflammation, the majority of anti-inflammatory drugs used in clinical trials did not demonstrate efficacy in preventing heart failure [42,43,44,45]. This lack of success may stem from the intricate immune cell cascade that generates multiple pro-inflammatory mediators, resulting in the limited effectiveness of anti-inflammatory treatments [46]. Several other cardiovascular diseases, such as hereditary structural cardiomyopathy and idiopathic dilated cardiomyopathy, are also associated with NLRP3 inflammasome activation [47]. The involvement of NLRP3 inflammasome in the advancement of myocardial dysfunction and non-ischemic dilated cardiomyopathy has been observed, primarily through cardiomyocyte apoptosis mediated by a cysteine asparaginase-1-dependent mechanism [48].
In conclusion, cardiovascular system diseases are associated with the activation of NLRP3 inflammasome. However, there is a lack of established clinical drugs specifically targeting NLRP3 inflammasome to manage cardiovascular diseases. Further investigations into the relationship between NLRP3 inflammasome inhibitory drugs and the progression of cardiovascular diseases are warranted to advance the development of pertinent clinical therapeutics.
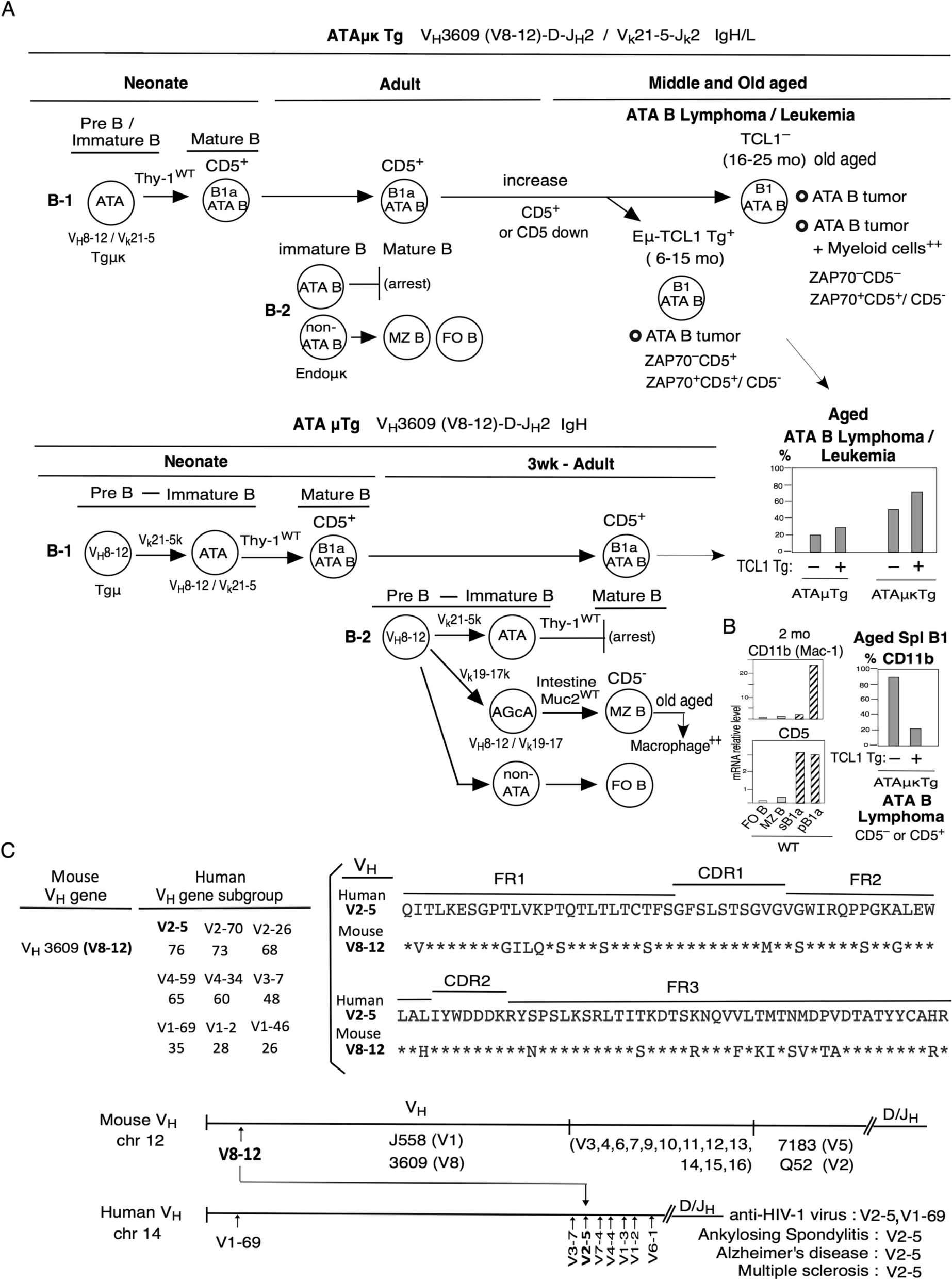
CancerThe relationship between cell senescence and cell carcinogenesis is bidirectional. On one hand, cellular senescence induces cell cycle arrest, which can reduce damage to cells during mitosis, such as cancerous cells caused by genetic mutations. Therefore, cellular senescence is an important barrier to prevent cell carcinogenesis to a certain extent [14]. On the other hand, some inflammatory factors like the NLRP3 inflammasome and some other chemokines like IL-6 and IL-8, produced by senescent cells create a conducive growth environment for neighboring cells in a precancerous state that can progress into tumor cells, accelerating their carcinogenesis process [49]. Under the dual effect of cell senescence, the fate of cells depends on the characteristics of the cells themselves and the different external stimuli that impact how cells respond to the senescent process. The developments of different cancers are presented in Fig. 4.
Fig. 4
Association of NLRP3 inflammasome with cancer progression. 5-HT, 5-hydroxytryptamine; ABHD5, α/β-Hydrolase domain-containing protein 5; CRC, colorectal cancer; EMT, epithelial-mesenchymal transition; IL-18, interleukin-18; IL-1β, interleukin-1β; LNCaP, lymph node carcinoma of the prostate; P2Y2R, P2Y purinergic receptor 2; PCa, prostate cell; PC-3, prostate cancer cell line; TAM, tumor-associated macrophage; TGF-β1, transforming growth factor-β1; TNF-α, tumor necrosis factor-α; TRIM59, tripartite motif-containing 59
Prostate cancerXu et al. demonstrated that NLRP3 inflammasome significantly enhanced the proliferation and migration of human prostate cancer cells, prostate cancer cell (PC) line 3 and lymph node carcinoma of the prostate (LNCaP) cells, but decreased the apoptotic ability of PCa cell lines by cell counting kit-8 (CCK-8), TdT-mediated dUTP nick end labeling (TUNEL) and Transwell assays [50]. Their further studies revealed that NLRP3 inflammatory vesicles promote malignant transformation of PCa through activation of caspase-1, providing new possible prognostic biomarkers and potential therapeutic targets for PCa [50]. The study by Zhao et al. also showed that chronic inflammation triggered by the NLRP3 inflammasome is one of the crucial factors leading to the occurrence and development of cancer. They also achieved the effect of directly using NLRP3 inhibitors on cells by interfering with the acetylation of NLRP3, that is, inhibiting the malignant progression of PCa [51].
However, while chronic inflammation caused by NLRP3 inflammasome promotes the occurrence and development of cancer, there is also evidence that these inflammatory factors can induce cell cycle arrest by promoting cell senescence to inhibition [52]. Studies supporting this view have shown that tumor development is inseparable from the microenvironment provided by tumor-associated macrophages (TAM). Re-educated TAM to adopt a pro-inflammatory and anti-tumor phenotype shifts the originally pro-cancer inflammatory factors into anti-cancer agents, thereby suppressing cancer development [52, 53].
The above findings underscore the role of chronic inflammation in PCa induction. Integrated with NLRP3 inflammasome research, targeted pharmacological interventions can alter cell phenotypes promoting cancer, inducing a positive shift towards cell senescence. Simultaneously, this intervention inhibits cancer onset and development, offering a novel treatment idea for PCa. The regulatory mechanism linked with NLRP3 may become a focal point for future research.
Lung cancerIn the study by Liang et al., it was observed that tripartite motif-containing 59 (TRIM59) released by tumors is expressed in exosomes. Through the regulation of the proteasomal degradation of Alpha–beta hydrolase domain-containing 5 (ABHD5), macrophages acquire tumor-promoting functions. These altered macrophages activate the NLRP3 inflammasome signaling pathway, leading to an upregulation in the secretion of IL-1β. This process promotes the formation of an inflammatory microenvironment and facilitates cancer metastasis to the lungs, ultimately contributing to the development of lung cancer [54]. In another experiment, researchers discovered the pivotal role of IL-1β in the development of inflammation-induced lung tumors. The mechanism involves IL-1β facilitating the proliferation and migration of non-small cell lung cancer cells by mediating the inhibition of microRNA-101, which has anti-tumor effects [55]. Additionally, data from a clinical trial suggest that canakinumab, a therapeutic agent targeting the IL-1β innate immune pathway, holds significant potential in reducing both the incidence and mortality of lung cancer [56]. Research on salidroside has revealed its inhibitory effects on lipopolysaccharide-induced proliferation and metastasis of non-small cell lung cancer. Salidroside achieves this by suppressing the activation of the NLRP3 inflammasome. These findings indicate that NLRP3 inflammasome-mediated inflammation plays a crucial role in the proliferation and metastasis of lung cancer. Furthermore, aging is implicated in the upregulation of various inflammatory factors, including the NLRP3 inflammasome [57].
Moreover, research has demonstrated a correlation between the occurrence and development of lung cancer and the concentration of IL-1β. Low concentrations of IL-1β have been shown to stimulate the production of anti-cancer factors and activate the body's inherent immune system against cancer. Conversely, elevated concentrations of IL-1β directly contribute to the proliferation, differentiation, and metastasis of cancer cells to other tissue cells [58]. Future investigations could delve into the intricate relationship between IL-1β levels and cancer progression, providing a theoretical basis for the development of drugs modulating NLRP3 inflammasome.
Breast cancerBreast cancer stands out as one of the most malignant tumors impacting women. Its pathogenesis intertwines with factors such as inflammation, age, estrogen levels, and breast tissue density. In the context of inflammation, the normal inflammatory processes become dysregulated during cellular aging, resulting in cellular damage and the prolonged release of pro-inflammatory factors. The accumulation of damaged cells within the tissue can precipitate cancerous transformations within the tissue landscape [59].
In an animal experiment in a mouse model of breast cancer, Wallenstein et al. demonstrated that the deletion of a human tumor suppressor gene (p53) in cancer cells induces TAMs to produce IL-1β, thereby promoting systemic inflammation and drive breast cancer metastasis [60]. Jin et al. reported that the NLRP3 inflammasome can be induced and activated by P2Y purinergic receptor 2 (P2Y2R) in metastatic breast cancer cells, enhancing tumor invasion and tumor growth, and contributing to tumor progression [61]. Moreover, anatomical experiments analyzing 53 breast tumor patients revealed a significant upregulation of gene expression in the NLRP3 inflammasome pathway in human breast tumor stroma compared to normal tissue stroma, suggesting an non-negligible role for this pathway in tumor progression [62]. However, the mechanism by which NLRP3 inflammasome regulate tumor progression is currently unknown.
The abnormal activation of the NLRP3 inflammasome, which may promote cancer occurrence, has spurred extensive clinical research on the development of NLRP3 inflammasome inhibitors. Among these, inhibitors targeting the NLRP3 inflammasome by blocking the IL-1 signaling pathway prove effective in breast cancer treatment. Inhibitors like anakinra and rilonacept have demonstrated promise in clinical trials [63]. Results from these trials indicate that these inhibitors significantly reduce tumor volume, inhibit growth, and alleviate symptoms and discomfort in patients. This introduces a new treatment choice for breast cancer patients, potentially enhancing their survival and quality of life.
In conclusion, inflammation emerges as a dominant force in promoting breast cancer occurrence, and NLRP3 inhibitory drugs, studied extensively to impede inflammasome activation, provide a new direction and hope for breast cancer treatment. However, further studies are necessary to confirm the safety and efficacy of these drugs in breast cancer treatment.
Colorectal cancerChronic inflammation stands out as a pivotal factor in the development of colitis-associated colorectal cancer. The neurotransmitter 5-hydroxytryptamine (5-HT) emerges as a promoter of NLRP3 inflammasome generation, initiating chronic intestinal inflammation. This sets in motion a positive feedback loop, as the NLRP3 inflammasome, in turn, mediates IL-1β maturation and catalyzes 5-HT biosynthesis by inducing tryptophan hydroxylase 1 synthesis, thereby elevating 5-HT levels in intestinal epithelial cells [64].
In the context of colorectal cancer, Wang et al. observed heightened NLRP3 expression in mesenchymal-like colon cancer cells (SW620). They demonstrated that TNF-α and transforming growth factor-β1 (TGF-β1) could upregulate NLRP3 expression in colon cancer epithelial cells, contributing to the epithelial-mesenchymal transition process—a critical facet of cancer cell metastasis. When NLRP3 was downregulated in colorectal cancer cells, a reduction in cell migration and invasion was noted [65]. However, using a colorectal cancer metastasis model to the liver, Maryse Dagenais et al. discovered that cancer-induced NLRP3 activation stimulates NK cell responses via IL-18, potentially enhancing tumor clearance [66].
The intricate relationship between NLRP3 inflammasome activation and cancer progression poses numerous areas for investigation. Key questions include whether NLRP3 inflammasome activation promotes cancer progression, its specific impact on various cancers, and the mechanisms through which inhibiting NLRP3 inflammasome activation mediates cancer progression—all warranting further exploration.
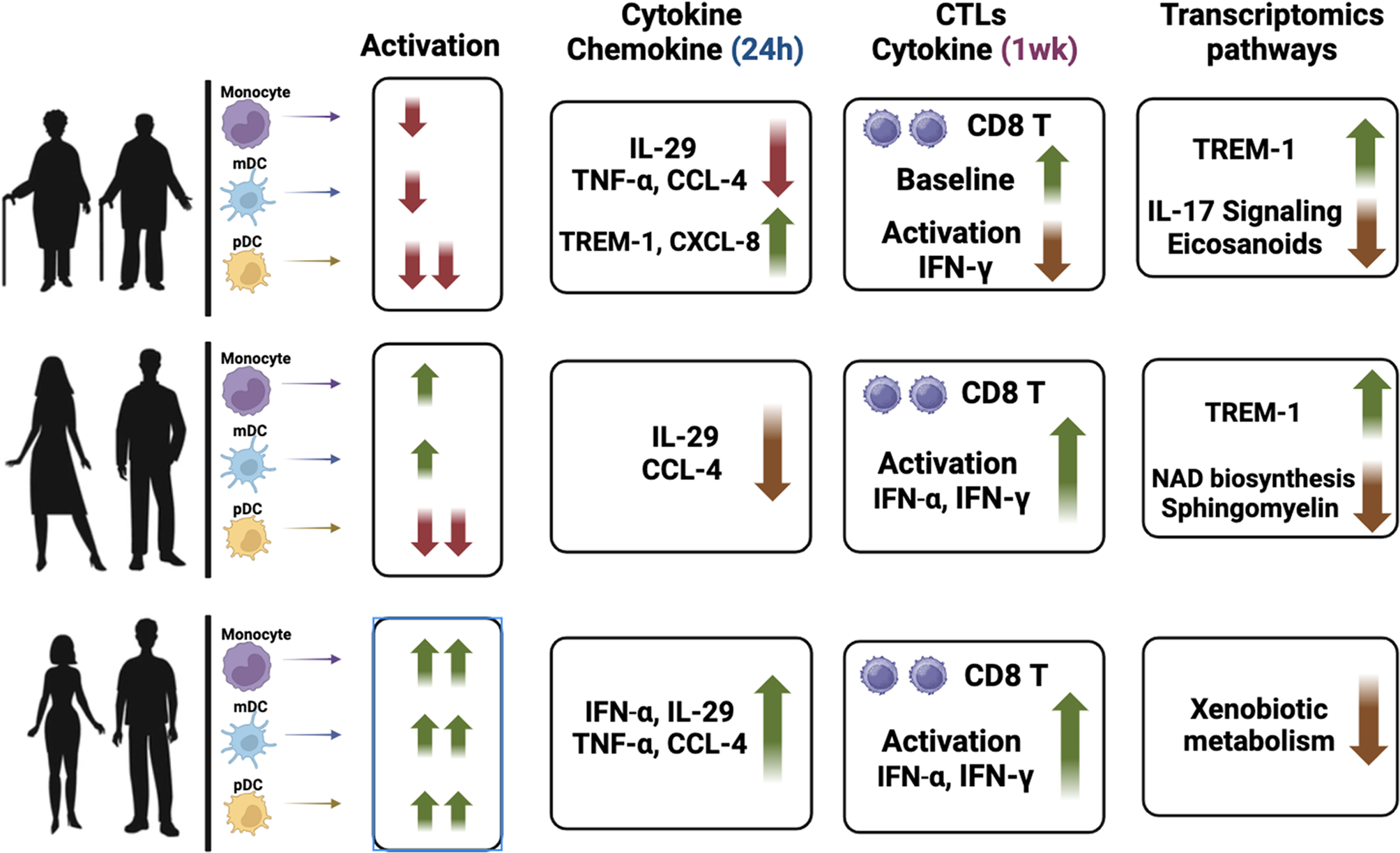
ArthritisThere is substantial evidence linking chronic or excessive inflammation to the development of arthritis [67]. Osteoarthritis, a prevalent degenerative joint disease affecting nearly any joint, exhibits elevated levels of DAMPs, notably basic calcium phosphate, in affected joints compared to normal joints. These DAMPs activate NLRP3, prompting the release of IL-1β and IL-18 from activated macrophages into the synovial fluid. This amplifies the inflammatory response cascade, culminating in chondrocyte tissue death and cartilage degeneration [68] (Fig. 5). In rheumatoid arthritis, an autoimmune disorder, TLR activation by DAMPs induces NLRP3 inflammasome expression and IL-1β release from macrophages [69]. Furthermore, stimulation of macrophages with citrulline-containing fibrinogen could indirectly stimulate NLRP3-induced IL-1β release through activation of TLR4 [70] (Fig. 5). Gouty arthritis is also an inflammatory disease in which severe pain is caused by the deposition of many monosodium urate crystals in the joints and surrounding tissues. In a drug experiment on resveratrol, the researchers revealed that resveratrol inhibited NLRP3 inflammasome activation by promoting mitophagy in a rat arthritis model, ultimately ameliorating arthritis symptoms [71] (Fig. 5).
Fig. 5
NLRP3 inflammasome in the progression of certain other aging-related diseases. BCP, basic calcium phosphate; caspase-1, cysteine-aspartic protease 1; CRP, C-reactive protein; DAMPs, damage-associated molecular patterns; IAPP, islet amyloid polypeptide; IL, interleukin; LPS, lipopolysaccharides; pro-IL-1β, pro-interleukin-1β; PDTC, pyrrolidine dithiocarbamate; PVN, paraventricular nucleus; SHR, spontaneously hypertensive rats; T2DM, type 2 diabetes mellitus; TLR4, toll-like receptor 4; TLRs, toll-like receptors; VEGF-A, vascular endothelial growth factor A; VSMCs, vascular smooth muscle cells
While extensive research confirms NLRP3 inflammasome involvement in the pathophysiology of arthritis and identifies potential inflammatory mechanisms, some interventions targeting these pathways have not transitioned successfully into clinical treatments. Further exploration is essential to grasp the potential simultaneous activation of multiple pathways driving joint inflammation, paving the way for more effective therapeutic strategies.
CataractAging constitutes a significant risk factor for the development of cataracts. Over time, the lens accumulates various harmful irritants, triggering chronic inflammation in the eye. The NLRP3 inflammasome is an indispensable factor in mediating inflammation, and its massive secretion can result in cellular self-damage, contributing to the gradual loss of lens transparency [72]. Clinical experiments conducted by Deng et al. demonstrated that cataracted eyes of 70 patients released inflammatory factors, including the NLRP3 inflammasome, through the induction of epithelial cell pyroptosis [73]. Similarly, Sun et al. observed significantly increased concentrations of key markers such as active caspase-1, IL-1β, and IL-18 in the capsule tissue of cataract patients [74].
Additional studies have highlighted the occurrence of cellular hypoxia and oxidative damage in various tissues with age. These processes induce the expression of vascular endothelial growth factor A (VEGF-A) in the retinal pigment epithelium. VEGF-A, in turn, induces ROS production, leading to the release of inflammatory mediators from the NLRP3 inflammasome, exacerbating cellular and tissue damage. This interaction establishes a detrimental cycle that intensifies inflammatory responses and tissue damage. Ultimately, the increasing number of damaged cells contributes to the gradual loss of lens transparency, leading to the development of cataracts [75]. In summary, cataract development is intricately linked to chronic inflammation-induced cell pyroptosis, resulting in reduced lens transparency (Fig. 5).
Osteoporosis (OP)OP is a systemic bone metabolic disease and a prevalent cause of illness and mortality among the elderly [76]. For a long time, it has been widely believed that OP is an inevitable result of aging. Aging and reduced estrogen levels drive low-grade inflammation in the body, and the production of pro-inflammatory cytokines affects the growth of osteoblasts and osteoclasts, ultimately stimulating the development of OP [77]. Notably, the latest research results show that OP is preventable and treatable, which depends on the discovery of novel mechanisms contributing to its development [78].
NLRP3 plays a dual role in bone metabolism (Fig. 5). On one hand, as the adaptor protein ASC is crucial in osteoblast development, the NLRP3 inflammasome and its associated proteins actively regulate bone growth and development [79]. On the other hand, overactivation of the NLRP3 inflammasome causes aging-related osteopenia. In OP mouse models, notable symptoms of granulocyte infiltration and significantly elevated IL-1β levels were observed [80]. Within osteocytes, the NLRP3 inflammasome inhibits osteoblast activation and accelerates osteoclast differentiation through the activation of IL-1β and IL-18, leading to OP.
Overall, the NLRP3 inflammasome exerts a more significant impact on osteoclasts than osteogenesis in osteocytes. Consequently, reducing the expression of NLRP3 in osteocytes emerges as a crucial strategy to decelerate the progression of OP [76]. Given the dual role of the NLRP3 inflammasome in OP pathogenesis, research and trials involving targeted therapy inhibitors warrant careful consideration.
DiabetesSterile inflammation emerges as a pivotal factor in the aging process, and accumulating evidence suggests its involvement in insulin resistance among older adults [81]. Diabetes, characterized by elevated blood sugar levels resulting from defects in insulin secretion or action, stands as one of the most prevalent metabolic diseases globally, accompanied by severe complications.
A key pathophysiological process in the development of T2DM is insulin resistance, closely entwined with inflammatory factors such as IL-1β, where the NLRP3 inflammasome assumes a central role [82]. Islet amyloid polypeptide (IAPP), or amyloid, does not form active amyloid aggregates in mice. Leveraging this characteristic, a transgenic mouse model overexpressing human IAPP was generated, revealing the induction of IL-1β production by macrophages in pancreatic islets in vivo. Another hallmark of T2DM is the accumulation of substantial amylin in the pancreas, which, upon uptake by macrophages and dendritic cells, activates the downstream NLRP3 inflammasome [83] (Fig. 5).
Moreover, metformin, the primary drug for T2DM treatment, exhibits its efficacy by reducing IL-1β levels through NLRP3 inflammasome inhibition, thereby alleviating symptoms in patients. This underscores the pivotal role of the NLRP3 inflammasome in driving the onset and progression of T2DM [84].
HypertensionHypertension is a prevalent condition, particularly affecting the middle-aged and elderly population, where aging stands out as a significant contributing factor. Chronic inflammation is recognized as a common mechanism associated with aging, and C-reactive protein, an inflammatory biomarker, consistently exhibits elevated concentrations with age in the absence of overt infection [85]. Studies indicate a higher plasma concentration of C-reactive protein in hypertensive patients compared to their normal counterparts, suggesting an association between hypertension pathogenesis and inflammation [86, 87].
Using spontaneously hypertensive rats (SHR) and primary active vascular smooth muscle cells (VSMCs) from hypertensive vascular smooth muscle cells, Sun et al. investigated that in SHR-derived VSMCs, increased histone acetylation promotes nuclear factor kappa-B (NF-κB) activation, subsequently activating the NLRP3 inflammasome. Intravenous injection of NLRP3-shRNA adenovirus can down-regulate NLRP3 protein, resulting in decreased expression of caspase-1 and IL-1β protein in SHR, leading to reduced blood pressure [88]. Furthermore, Zhu et al. observed persistent elevation of NF-κB activation, the initiator of the NLRP3 inflammasome, in hypertensive patients [89]. Targeted therapies for hypertension, such as NF-κB inhibitors like IMD-0354, have proven effective in preventing the increase in right ventricular pressure [90]. Another example is pyrrolidine dithiocarbamate, a compound that inhibits NF-κB and can be infused into the hypothalamic paraventricular nucleus to block high salt by inhibiting the NLRP3 inflammasome and caspase-1, thereby inducing hypertension development [91]. This supplies therapeutic directions for
留言 (0)