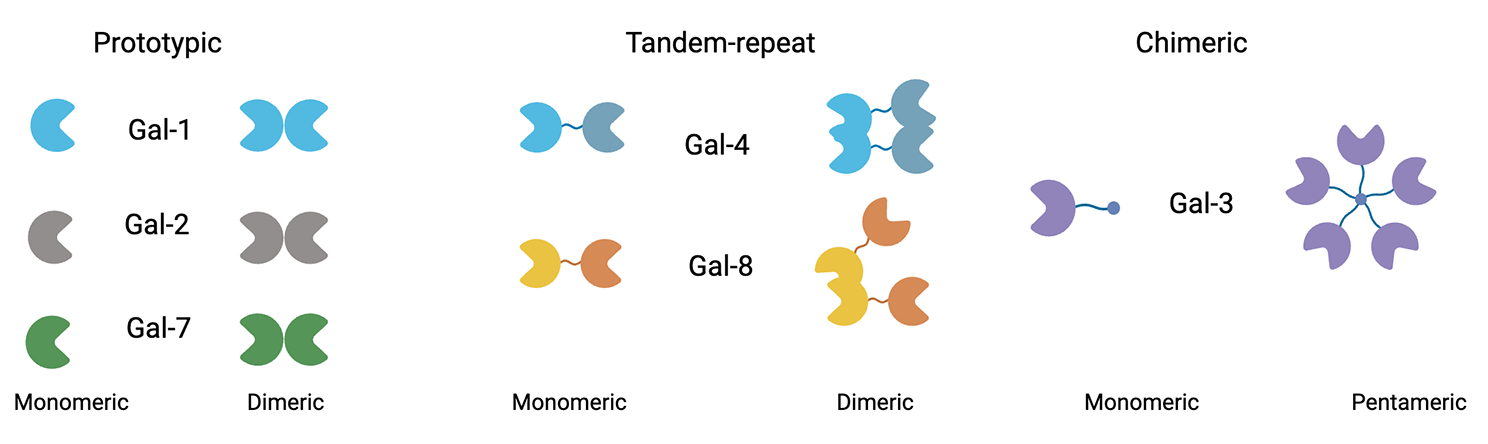
Animals
Animal experiments were approved by the Animal Welfare and Ethics Committee of School of Stomatology, the Fourth Military Medical University following the “Guidelines for the Care and Use of Laboratory Animals”. Wild-type C57BL/6 mice were obtained from the Laboratory Animal Research Center of the Fourth Military Medical University, which were randomly grouped and used for in vivo studies. All mice were housed in a 12-h dark–light cycle at 22 ± 2 °C and 40% humidity and all mice were allowed access to a standard diet. The sample size (n = 6) was decided by “resource equation” method. The confounders were not controlled. No criteria were set to include or exclude animals. The 7-day-old mice were injected subcutaneously with neomycin (dissolve in sterile saline) once per day for five consecutive days at a dose of 200 mg/kg, while the control group was injected with the same amount of sterile saline into the same region without neomycin. The detailed protocol for neomycin administration was given previously [43]. Two days later, exosomes (20 μg in 10 μl PBS) were injected into the mice through the round window niche (RWN) injection, while the control group was injected with the same amount of PBS into the same region without exosomes. Autophagy inhibition in mice was achieved through intraperitoneal (IP) injection of 3-MA at a dose of 30 mg/kg, administrated three times at 24 h before, 2 h before and immediately after RWN treatment. The hearing threshold was evaluated by ABR measurement at P28. After ABR measurement, cochlear tissues were collected for immunofluorescence staining. CAG-RFP-EGFP-LC3 transgenic mice (Jackson Laboratory, 027139) obtained from the Jackson Laboratory were used to test the level of autophagy in the cochlear hair cells. ABR measurement and assessment were performed in a blinded manner. For other experiments, investigators were not blinded since they needed to perform these experiments with different treatments.
Cell culture, tissue culture and reagents
Umbilical cord mesenchymal stem cells (UC-MSCs) were purchased from Procell (CP-CL11, China) and cultured in αmodified Eagle medium (α-MEM) (Gibco, USA) supplemented with 10% FBS (Sijiqing, China), 2 mM L-glutamine (Invitrogen, USA), 100 U/mL penicillin and 100 U/mL streptomycin (Invitrogen, USA). Cochleae were dissected from P1-3 mice and cultured as previously reported [44]. Briefly, cochleae were dissected and cleaned of surrounding tissue and bone in Hank’s Balanced Salt Solution (Solabio, H1045; China). The cochlear explants were attached to a glass coverslip coated with collagen solution (10X Basal Medium Eagle (BME; Sigma, B9638), 2% sodium carbonate (Sigma, S7795), collagen gel type I (Corning, 354,236), in a 1:1:9 ratio) [45]. The explants were incubated in DMEM/F12 medium supplemented with N2/B27 (Gibco, 17,502,048/17504044, USA) and ampicillin at 37 °C with 5% CO2 overnight prior to each treatment to stabilize them. Neomycin sulfate (Sigma-Aldrich, USA, N6386) was used to damage hair cells at a concentration of 0.5 mM for 24 h. After neomycin was removed, the tissues were treated with exosomes (30 μg/ml for 24 h) or the same amount of PBS. HEI-OC1 cells were cultured at 33 °C with 10% CO2 in high-glucose Dulbecco's Eagle's medium (DMEM) containing 10% FBS without antibiotics. When cells reached a suitable density, neomycin was added to the medium at a final concentration of 2 mM for 24 h to damage the HEI-OC1 cells. After neomycin was removed, the cells were treated with exosomes (30 μg/ml for 24 h) or the same amount of PBS. Chloroquine (HY-17589A), 3-MA (HY-19312), Dynasore (HY-13863), and Cytochalasin D (HY-N6682) were purchased from MCE (USA).
Western blot
Cells and tissue samples were lysed with RIPA buffer (Beyotime, China) containing protease inhibitor. Protein quantification was performed using a BCA assay (Beyotime, China). Extracted proteins (30 µg) were separated by SDS–polyacrylamide gel electrophoresis (PAGE) and transferred onto PVDF membranes. The membranes were blocked with 5% bovine serum albumin for 1 h at room temperature, followed by incubation with primary antibodies overnight at 4 °C. After washing with Tris-buffered saline-Tween three times, the membranes were incubated with secondary antibodies for 2 h at room temperature. The following primary antibodies were used: Alix (1:1000; Cell Signaling, 92,880, USA), CD9 (1:1000; Abcam, ab236630, USA), CD63 (1:1000; Santa Cruz Biotechnology, sc-5275, USA), CD81 (1:1000; Santa Cruz Biotechnology, sc-9158, USA), GAPDH (1:5000; CWBIO, CW0100, China), Cleaved Caspase-3 (1:1000; Cell Signaling, 9661 s, USA), LC3A/B (1:1000; Cell Signaling 12,741, USA), SQSTMQ/p62 (1:1000; HUABIO HA721171, China), BECN1 (1:1000; Cell Signaling 3495S, USA), CAV(1:1000; Abcam, ab133484, USA), EEA1(1:1000; Abcam, ab109110, USA).
Isolation and characterization of exosomes
Cell supernatant was first centrifuged at 800 × g for 10 min to remove cells or cell debris. The supernatant was then centrifuged at 16,000 × g for 30 min to remove microvesicles. Next, the supernatant was ultracentrifuged at 150,000 × g for 70 min at 4℃, washed with PBS, and purified by ultracentrifugation at 150,000 × g for additional 70 min. Exosomes derived from MSCs were collected from the bottom of the tube and resuspended in sterile PBS for further use. The protein concertation was measured by BCA kit (Beyotime). The size distribution of exosomes was analyzed using nanoparticle tracking analysis (NTA) with Zeta View PMX 110 (Particle Metrix) and corresponding software, Zeta View 8.04.02. Exosomes were also observed directly under a transmission electron microscopy (TECNAI Spirit, FEI).
Internalization of exosomes into hair cells in vitro
The cochlear explants were attached to a glass coverslip coated with collagen solution and incubated in DMEM/F12 medium supplemented with N2/B27 and ampicillin at 37 °C with 5% CO2 overnight to stabilize them before each treatment. Exosomes were pre-labeled with the PKH26 Red Fluorescent Cell Linker Kit (Sigma-Aldrich, USA) according to the manufacturer’s instructions and washed in PBS with ultracentrifugation at 150,000 × g for 70 min. PKH26-labeled exosomes at a concentration of 20 μg/ml were then co-cultured with cochlear explants for 8 h or co-cultured with HEI-OC1 for 1 h. After fixation with 4% paraformaldehyde for 20 min at 4 °C, the explants were used for further immunofluorescence staining.
Immunofluorescence
The samples were fixed in 4% paraformaldehyde (PFA) for 20 min and permeabilized with 0.5% Triton X-100 for 15 min. After washing with PBS, the samples were incubated with primary antibodies at 4 °C overnight, followed by treatment with Cy-3- or FITC-conjugated IgG secondary antibody (1:200; Jackson, USA) for 2 h at room temperature. Cell nuclei were counter-stained with Hoechst 33,342 for 5 min at room temperature. For autophagy flux assay, HEI-OC1 cells were infected with adenoviruses expressing mRFP-GFP-LC3 (MOI 25, Genechem, China) for 72 h and then treated with exosomes and/or Chloroquine (CQ). Images were obtained using the laser scanning confocal microscope (Olympus or Nikon) and quantified. The following primary and secondary antibodies were used in the immunofluorescence studies: Myosin7a (1:400; Proteus biosciences, 25–6790, USA), Cleaved Caspase-3 (1:1000; Cell Signaling, 9661 s, USA), EEA1(1:100; Abcam, ab109110, USA), FITC-conjugated goat anti-rabbit IgG (1:200, Jackson, 111–095-003, USA), and Cy3-conjugated goat anti-rabbit IgG (1:200, Jackson, 111–165-003, USA). Rhodamine-phalloidin (Cytoskeleton, PHDR1, USA), Mito-SOX Red (Invitrogen, M36008, USA), and TUNEL kit (Beyotime, C1090, China) was used to label or measure F-actin, ROS levels and apoptosis according to the manufacturer’s instructions.
siRNA transfection
HEI-OC1 cells were transfected with siRNA (RiBOBIO) targeting ATG5 using Advanced DNA RNA Transfection Reagent (Zeta Life) according to the manufacturer’s instructions. Next, at 48 h after siRNA transfection, the efficiency of knockdown was determined by western blotting. Subsequent treatments on transfected cells were performed 48 h after transfection.
Gene expression analysis by real-time qRT-PCR
Total RNA was extracted with TRIzol reagent (Invitrogen, USA), and one microgram of total RNA was reverse transcribed into cDNA with a Prime Script RT reagent kit (TaKaRa, Japan) by the thermal cycler (C1000 Thermal Cycler, Bio-Rad, USA). Real-time RT-PCR was performed with SYBR Green dye and Taq polymerase (TaKaRa, Japan) by the CFX96TM Real-time RT-PCR System (C1000 Touch Thermal Cycler, Bio-Rad, USA). Gene expression was normalized to an internal control GAPDH. The primer sequences are shown in Table 1.
Table 1 Primers for real-time PCRFlow cytometry
Annexin V and PI assays were conducted to measure the apoptosis of HEI-OC1 cells using the Annexin V-FITC/PI Apoptosis Detection Kit (BD Biosciences, 556,547, USA) according to the manufacturer’s instructions. The cells were collected and resuspended in 100 μL of binding buffer, and stained with 5 μL of FITC labelled Annexin V and 5 μL of PI 15 min at RT (25 °C) in the dark. Add 400 µl of binding buffer to each tube. The stained cells were then analyzed by flow cytometry (Beckman-Coulter, USA).
Electron microscopy
Cochleae and HEI-OC1 cells were collected and immediately fixed in 2.5% glutaraldehyde for 24 h and then in 1% osmic acid for 2 h, dehydrated with acetone, and embedded in Epon 812. The ultrathin sections were stained with alcoholic uranyl acetate and lead citrate, washed gently with distilled water, and observed with transmission electron microscope (TECNAI Spirit, FEI).
ABR test
Under light anesthesia with sodium pentobarbital (40 mg/kg), the reference, ground, and active needle electrodes were inserted beneath the skin of the post-measured auricle, the sacrococcygeal region, and the calvaria of each mouse, respectively. ABR was measured using the Tucker-Davis Technology RZ6 system (TuckerDavies Technologies, Gainesville, FL, USA) at 4, 8, 16, 24, and 32 kHz, respectively. ABR waves I and II were monitored to assess thresholds.
Statistical analysis
Data were expressed as mean ± s.d as indicated. Comparisons between two groups were performed by Student’s t-test, and multiple group comparisons were performed by one-way ANOVA. Tukey’s correction was used when multiple comparisons were performed. P values less than 0.05 were considered statistically significant. Graphs and statistical analysis were performed using GraphPad Prism (GraphPad Software 7.0, USA). No animals or data points were excluded.






留言 (0)