
記住我
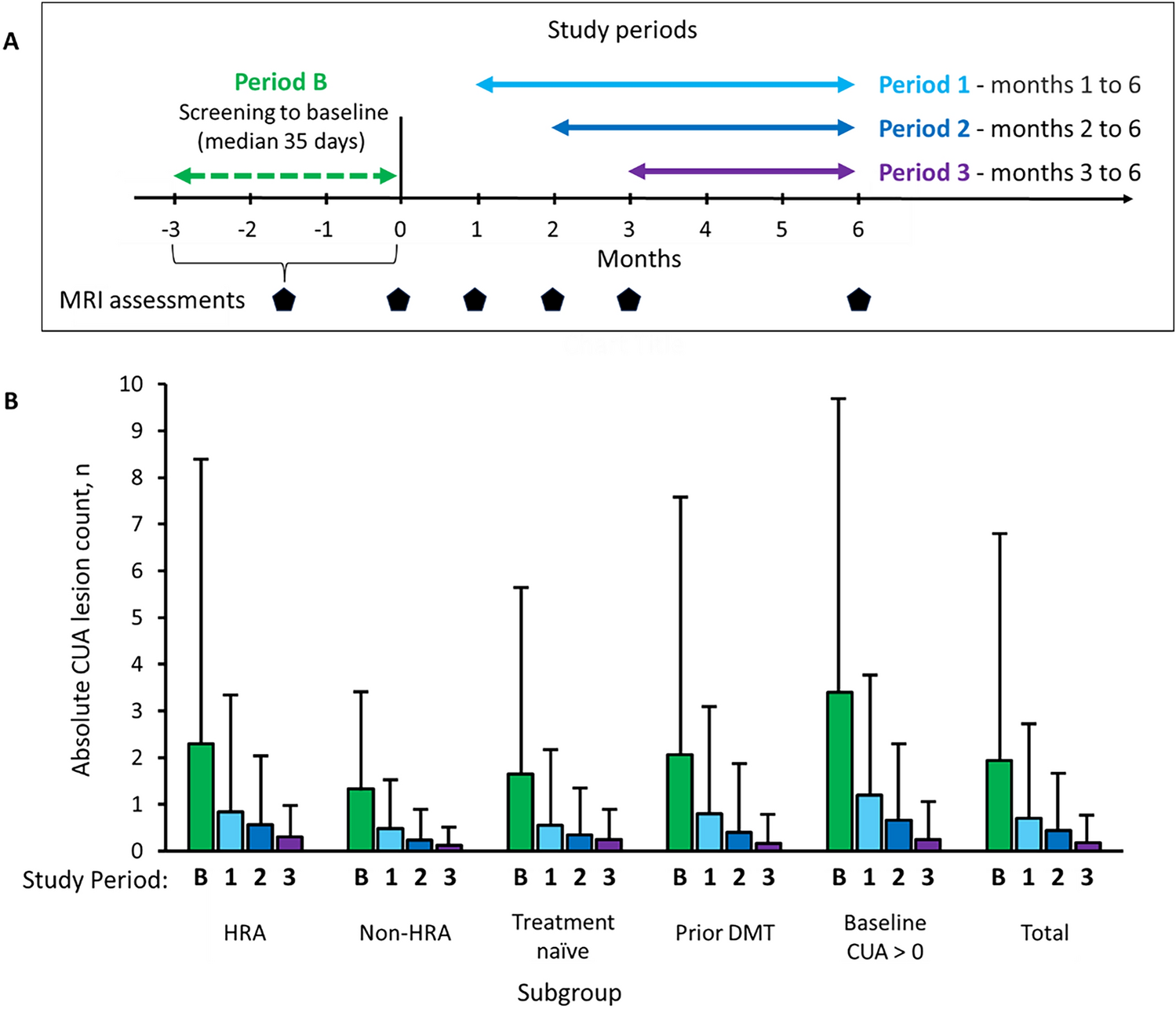
This was a Phase I, double blind, randomized (1:1), two-way cross-over, single dose, placebo-controlled study, in healthy male CCK-4 sensitive participants, comparing treatment with BI 1358894 to placebo (NCT03904576). The study took place at one site, PRA Health Sciences in the Netherlands. Treatments consisted of oral BI 1358894 100 mg (four 25-mg tablets) or oral matching placebo, both administered in the fed state (high fat/caloric meal 30 min before drug administration) and 5 hours (h) prior to the challenge agent, intravenous (IV) CCK-4 acetate 50 µg, which was administered at 13:00. Consistent timing of BI 1358894 and CCK-4 administration was maintained to avoid confounding factors relating to effects of circadian rhythms on data output. Treatment administration 5 h prior to the challenge agent was based on prior clinical data to ensure intended plasma concentrations of BI 1358894 were reached in > 95 % of participants. Participants were randomly allocated to one of two treatment sequences (BI 1358894 followed by placebo or placebo followed by BI 1358894), with a washout period of ≥ 17 days between treatments (Fig. 1). Randomization was performed by the sponsor using a validated system that was verified by an independent statistician. The block size was four and the randomization list was generated using a validated pseudo-random number generator and a supplied seed number such that the resulting allocation was both reproducible and non-predictable. The randomization list contained additional blocks to allow for participant replacement. Participants, investigators and persons directly involved in the conduct of the trial were blinded to trial treatments.
Fig. 1
Study design. *Preselected CCK-4 sensitive healthy volunteers were randomized to one of two treatment sequences: oral BI 1358894 100 mg in the fed state followed by oral placebo in the fed state, or vice versa (washout period ≥17 days). Treatments were administered 5 h prior to IV CCK-4 (50 µg). CCK-4 cholecystokinin-tetrapeptide, IV intravenous, QD once daily, R randomization, S screening
The PSS and EVAS tests were performed prior to study initiation (CCK-4 screening visit); additional tests took place on Day 1, at the time of first treatment (BI 1358894 or placebo; time = 0 h), then at 4 h, 4:45 h, 5:05 h, 5:10 h, 5:20 h, and 5:30 h; Day 9 and Days 35–40 (end of study), with scores calculated for each time point. The STAI testing was carried out on Day 1 at 4:45 h posttreatment, Day 9 and Days 35–40 (end of study). Adrenocorticotropic hormone/cortisol samples were taken at screening, then on Day 1 at 4:45 h, 5:05 h, 5:10 h, 5:20 h, 5:30 h, 6:00 h, 7:00 h, 8:00 h, and 10:00 h. For each patient, the maximum concentration of the analyte (Emax) and the area under the effect–time curve of the analyte in plasma over the time interval 4:45 h to 10:00 h (AUEC4.75–10) was assessed and the mean change from baseline in plasma ACTH and cortisol was calculated for each time point.
2.2 ParticipantsEligible participants were CCK-4 sensitive, healthy male volunteers, aged ≥ 18–65 years, with a body mass index (BMI) of 18.5–29.9 kg/m2. This study was limited to male participants as reproductive toxicology studies had not been performed at the time of this study. Sufficient CCK-4 sensitivity was defined as achievement of a sudden onset of anxiety and panic along with presence of at least 4 symptoms in the PSS with either a score of ≥ 2 on PSS item 15 (Anxiety and Fear for Apprehension) or achievement of a total PSS sum intensity score > 20 after IV CCK-4.
Key exclusion criteria included any finding deviating from normal in the medical examination that was considered to be clinically relevant, any evidence of a concomitant disease deemed clinically relevant by the investigator, disease of the central nervous system, being a smoker (more than 5 cigarettes or 3 pipes per day), drug abuse or a positive drug screening, inability to comply with the dietary regimen of the trial site, and any suicidal ideation of type 2–5 on the Columbia-Suicide Severity Rating Scale (C-SSRS) in the last 12 months.
2.3 Endpoints and Assessments2.3.1 Pharmacodynamic EndpointsThe primary endpoint was maximum change from baseline of the PSS sum intensity score after CCK-4 injection. Further endpoints of interest were: time to maximum change from baseline in PSS sum intensity score, maximum PSS sum intensity score, PSS total number of symptoms, change from baseline in the PSS total number of symptoms, change from baseline in EVAS score, time to maximum change from baseline in EVAS score, maximum EVAS score, the value of each STAI for state anxiety (form Y1) before CCK-4 injection (Epre), and plasma levels of ACTH and serum levels of cortisol following CCK-4 injection (maximum concentration, AUEC of plasma/serum ACTH/cortisol concentrations over the time interval 4.45 h to 10 h [AUEC4.75–10] and time to maximum concentration of the analyte in plasma/serum [tmax]). The change from baseline in PSS and EVAS scores were defined as the score values after CCK-4 injection at each time point minus the last score value before CCK-4 injection.
The PSS is a patient-reported outcome measurement tool derived from the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition, Revised (DSM-5-R), which is designed to assess CCK-4-induced panic symptoms in patients [14]. The PSS lists 18 symptoms to assess CCK-4-induced panic symptoms, including 4 somatic symptoms and 4 anxiety symptoms. Key symptoms are measured on a 5-point scale from 0 (not present) to 4 (severe) and a composite score is determined by summing the number and intensity rating of symptoms for each subject. These key symptoms include: dyspnea, palpitations/rapid heartbeat, sweating, faintness, unsteady feeling, dizziness, shaking/trembling, nausea, abdominal distress, choking, chest pain/discomfort, paresthesia, hot flushes/cold chills, feeling unreal/detached, anxiety/fear/apprehension [14].
The EVAS, an adaptation of the visual analog score (VAS), provides a subjective measure of current general emotional status of the participant using a 100-mm long horizontal line with verbal descriptors or word anchors at each end to express the extremes of each feeling [30]. Participants marked a position on the line that ranged between 6 facial expressions (smiling face to crying face), in accordance with their emotional status. This position was measured in mm from 0 mm (no symptoms) to 100 mm (severe symptoms).
The STAI is used to distinguish state anxiety (a temporary condition) from trait anxiety (a general tendency to perceive saturations as threatening). Participants rate themselves on a scale of 1–4 for 20 questions to evaluate their level of anxiety. Higher scores indicate greater levels of anxiety. Within this trial setting, as the STAI assessment was conducted before the CCK-4 challenge, the measure was considered to be an indicator of potential anticipatory anxiety in expectation of the response to CCK-4 [31].
2.3.2 Pharmacokinetic EndpointsPharmacokinetics were evaluated as further endpoints and were calculated for BI 1358894 and included maximum measured concentration of the analyte in plasma (Cmax), tmax (plasma), area under the concentration-time curve (AUC) of the analyte in plasma over the interval from 0 to the last quantifiable data point (AUC0–tz), AUC4.75–5.5 of the analyte, and the terminal half-life of the analyte in plasma (t1/2).
2.3.3 Safety and Tolerability EndpointsThe safety and tolerability of BI 1358894 was assessed based on adverse events (AEs; including clinically relevant findings from the physical and neurological examinations); safety laboratory tests; 12 lead electrocardiogram (ECG) measurements; continuous ECG monitoring; vital signs (blood pressure, pulse); and suicidality assessment (C-SSRS).
The provoked panic symptoms in response to CCK-4 injection (as documented by the PSS score) were considered pharmacodynamic parameters. As they were an expected and desired response in the context of this trial (even though they constitute an untoward medical occurrence), they were not recorded as AEs. All other observations not included on the PSS questionnaire that occurred during the AE reporting time period were documented as AEs.
2.4 Ethical ConsiderationsThe trial was carried out in compliance with the clinical trial protocol, which was in accordance with the principles of the Declaration of Helsinki [32], the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use Good Clinical Practice (ICH GCP) guidelines, applicable regulatory requirements and Boehringer Ingelheim standard operating procedures. All participants provided informed written consent in accordance with ICH GCP and local procedures. The study protocol was reviewed and approved by the local independent ethics committee (Medisch Ethische Toetsings Commissie van de Stichting Beoordeling Ethiek Bio-Medisch Onderzoek, Assen, The Netherlands) on April 10, 2019.
2.5 Statistical AnalysisAll pharmacodynamic, pharmacokinetic, and safety endpoints were assessed descriptively. The adjusted mean maximum change from baseline in respective measures of PSS (sum intensity score) and EVAS (total score) per patient, and the Epre for STAI total score were assessed using a linear mixed-effects model. Covariates included effects for treatment, period, and subject, in addition to period baseline and subject baseline. Adjusted means, as well as two-sided 90 % confidence intervals (CI) were calculated. The change in plasma ACTH and serum cortisol following CCK-4 stimulation were compared to baseline levels by examining Emax and AUEC4.75–10 within each treatment group.
Sample size was determined based on the probability of observing a meaningful reduction in PSS sum intensity score after a single dose of BI 1358894 when compared with placebo. A reduction of at least 20% in PSS sum intensity score relative to placebo was considered meaningful. For these calculations, the following assumptions were made: the standard deviation (SD) of the maximum change from baseline of the treatment contrast of PSS sum intensity score relative to placebo is 20%; the maximum change from baseline for PSS sum intensity score relative to placebo is 0% after the placebo treatment. Based on these calculations, a sample size of 20, with at least 15 evaluable patients, was considered sufficient to achieve the aims of this exploratory study.
With respect to missing data, for PSS and EVAS, if the period baseline value was missing in treatment period 1, it was imputed by the corresponding value measured in treatment period 2. If the period baseline value was missing in treatment period 2, it was imputed by the corresponding value measured in treatment period 1. For the endpoint STAI, missing values were not imputed.
留言 (0)