
記住我
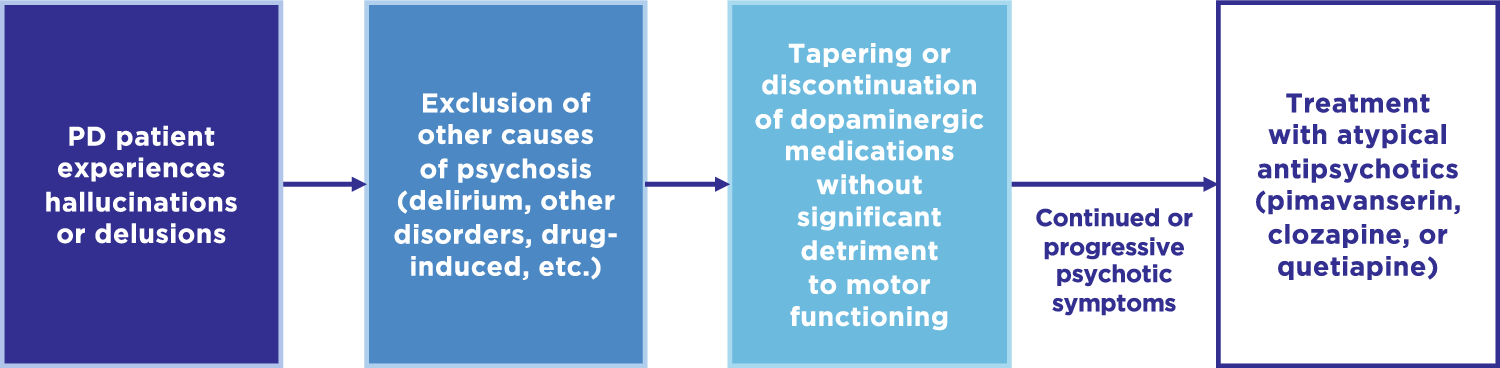
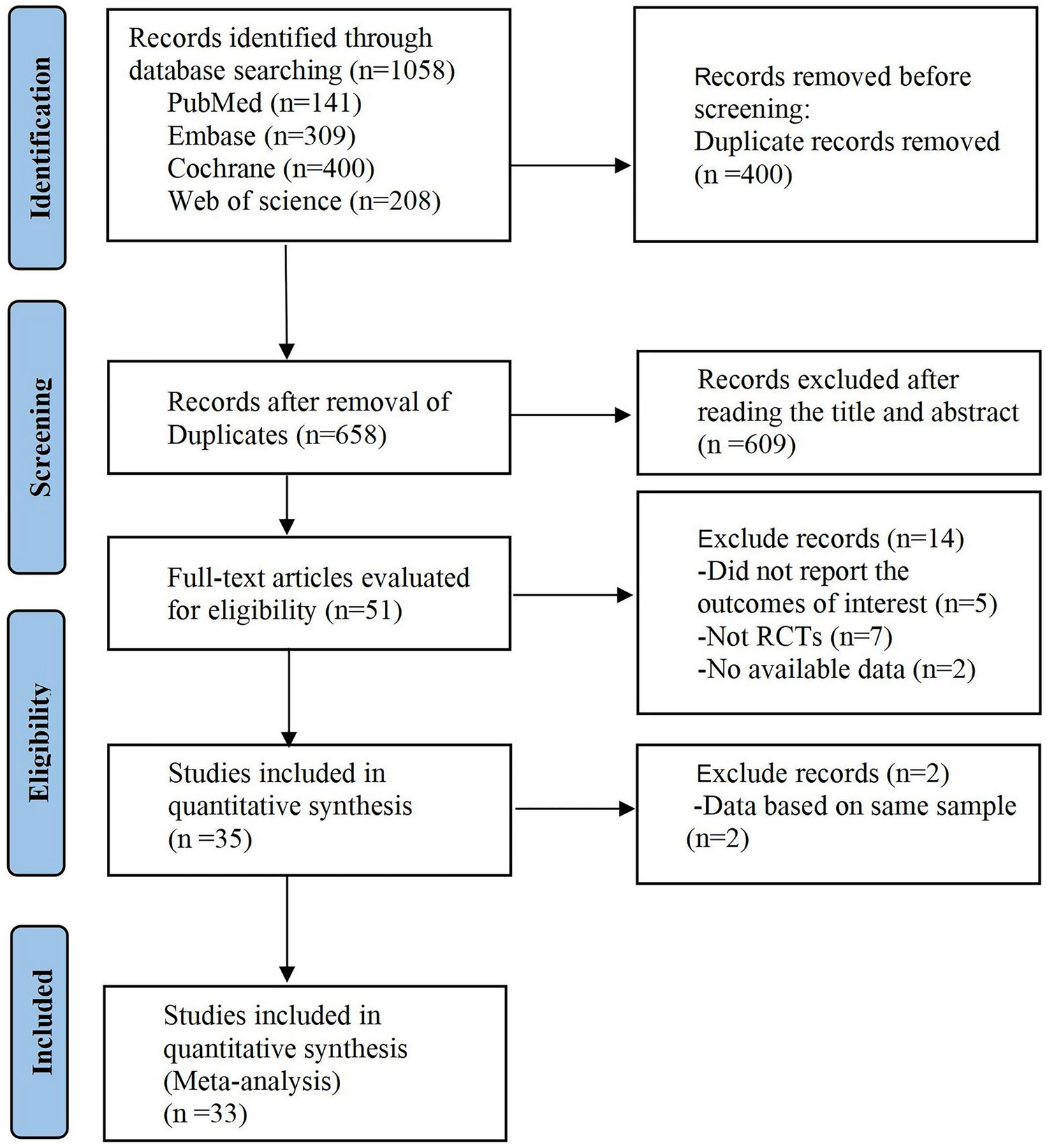
As noted above, before a diagnosis of PDP can be made, alternative causes of psychosis must be excluded [21]. Although differentiating PDP from other neurodegenerative disorders (e.g., Lewy body dementia, Alzheimer’s disease) can be challenging due to comorbidity of neuropathology, assessing dementia, including its timing of onset and evolution, in relation to the onset of psychosis can often be a distinguishing factor. Other psychosis-associated psychiatric disorders (e.g., mood disorders, schizophrenia, etc.) can be more easily differentiated from PDP by their onset and longitudinal development, or types of psychotic symptoms. Finally, delirium- or substance-associated psychosis should be considered, and managed accordingly, before the diagnosis and treatment for PDP [21, 22]. If psychotic symptoms persist despite resolving any delirium or discontinuing psychosis-inducing medications, and other psychosis-associated disorders have been ruled out, management and treatment of PDP may be considered. The standard PDP management paradigm usually involves tapering or discontinuation of dopaminergic medications, followed by treatment with atypical antipsychotics (Fig. 1).
Fig. 1
Current treatment algorithm for Parkinson’s disease psychosis based on guidelines and recommendations. PD Parkinson’s disease
3.1 Tapering or Discontinuation of Antiparkinsonian MedicationsMost medications used to manage motor symptoms in PD have been associated with hallucinations or delusions [22, 25]. A retrospective study of 52 PD patients without dementia showed that a high daily dose of levodopa (>750 mg daily equivalent) is associated with a sense of presence in PDP (OR 1.7, 95% CI 1.1–2.7; p = 0.029) [26]; however, an earlier case series of five patients with PDP who were treated with increasing amounts of levodopa found no association with hallucinations [27], although the differences seen in these two studies could be attributed to differences in sample sizes or inclusion criteria. Patients with PD who receive dopamine agonists (DAs) are reportedly more likely to experience symptoms of psychosis than patients receiving levodopa, when compared with untreated control patients with PD [28]. A meta-analysis of 25 trials evaluating DAs versus placebo or levodopa between 1990 and 2007 found that DAs are approximately five times more likely to be associated with hallucinations than placebo and twice more likely than levodopa [29]. Additionally, recent studies have found an association between the use of catechol-o-methyl-transferase (COMT) inhibitors and anticholinergic agents with PDP [30].
Current guidelines agree that when PDP symptoms become bothersome, antiparkinsonian medications should be optimized to diminish psychotic symptoms without significantly deteriorating motor function [22, 31,32,33,34]. However, there is no consensus on the order in which antiparkinsonian medications should be altered. Nevertheless, recent reviews have suggested that anticholinergic agents and DAs should be reduced, or stopped, first, followed by COMT and monoamine oxidase-B (MAO-B) inhibitors, leaving the alteration of levodopa formulations as a last resort [22, 31, 34].
If psychotic symptoms persist despite the adjustment of antiparkinsonian medications, in PDP patients with concomitant dementia or who are otherwise cognitively impaired, cholinesterase inhibitors (eg, rivastigmine, donepezil, and galantamine) should be considered [34, 35]. Cholinesterase inhibitors decrease the breakdown of acetylcholine, which allows for increased availability for acetylcholine in neuromuscular junctions to activate receptors associated with cognitive function. These agents are often approved for the treatment of dementia associated with PD and/or Alzheimer’s disease and many have demonstrated activity against psychosis-related symptoms such as hallucinations [34]. For example, a recent meta-analysis of 34 randomized clinical trials evaluating the use of cholinesterase inhibitors to treat symptoms of psychosis in patients with Alzheimer’s disease and PD showed an association between cholinesterase inhibitors and improvements in both delusions and hallucinations for both Alzheimer’s disease and PD patients [36].
If antipsychotic symptoms still persist after these routes, it is then recommended to consider initiating therapy with antipsychotic drugs [22, 31, 33, 34].
3.2 Typical and Atypical AntipsychoticsTypical first-generation antipsychotics (e.g., haloperidol, prolixin, etc.) alleviate psychotic symptoms primarily by strongly inhibiting the D2 receptor [37]. These agents can be used to treat most psychotic disorders (schizophrenia, schizoaffective disorder, etc.), however they can severely worsen motor symptoms and are associated with increased mortality. A recent retrospective matched-cohort study found that patients exposed to typical antipsychotics had a significant increase in mortality over those patients not treated with an antipsychotic. Additionally, patients treated with typical antipsychotics had a 62% higher risk of mortality than those treated with atypical psychotics [38]. Typical antipsychotics are also the most common cause of drug-induced Parkinsonism and other extrapyramidal adverse effects in patients without PD, with approximately 80% of patients receiving typical antipsychotics experiencing more than one type of extrapyramidal adverse effect [39, 40]. All current guidelines and recommendations advise against using typical antipsychotics to manage PDP [22, 31, 33, 34]. Moreover, the American Geriatrics Society (AGS) specifically recommends avoiding all typical antipsychotics for elderly PD patients via the 2019 Updated AGS Beers Criteria® for Potentially Inappropriate Medication Use in Older Adults [41].
Atypical antipsychotics can often inhibit the D2 receptor, but with a lower affinity than typical antipsychotics, and are serotonin 5HT2 receptor antagonists. While atypical antipsychotics such as risperidone [42,43,44], olanzapine [45, 46], and ziprasidone [47, 48] have shown promising antipsychotic efficacy in small PDP studies, they also worsened motor function. In a retrospective analysis by Weintraub and colleagues, atypical antipsychotics were associated with increased risk for mortality among patients with PDP, but to a lesser extent than typical antipsychotics [38]. Guidelines and recommendations, including the AGS Beers Criteria®, advise against using most atypical antipsychotics, for PDP, except for three agents, i.e. clozapine, quetiapine, and pimavanserin.
3.3 ClozapineClozapine was the first atypical antipsychotic shown to provide promising antipsychotic efficacy without significant worsening of PD-associated motor symptoms in a small cohort of PDP case studies [49]. Clozapine has since been evaluated in randomized placebo-controlled or active treatment comparator clinical trials for treating PDP-associated hallucinations and delusions (H&D), with clearly demonstrated efficacy (Table 3).
Table 3 Evidence of clinical efficacy with clozapine in select placebo-controlled or active comparator randomized trials in Parkinson’s disease psychosisHowever, clozapine is associated with a high incidence of drowsiness (up to 53%) and orthostatic hypotension (up to 19%) when used to treat PDP [50, 51]. In other disease settings, clozapine has also been linked to a risk of agranulocytosis [52] and metabolic syndrome [53]. Additionally, more than 90% of patients receiving clozapine may experience drug-induced sialorrhea, either diurnally or nocturnally, which can negatively impact patient quality of life [54, 55]. In a randomized study evaluating the use of clozapine (12.5–50 mg/day, mean dose of 35.8 mg/day) versus placebo in 60 patients with PDP, a numerically higher rate of worsening of parkinsonism was observed with clozapine compared with placebo (13% vs. 4%); however, most worsening of motor function was mild and/or transient and may have occurred due to higher doses of clozapine (only 3 of 14 cases of worsening parkinsonism were considered a large [>10%] decrease based on the Schwab and England score, and 2 of the 3 cases received 50 mg/day dosing of clozapine) [51]. It should be noted that the dose of clozapine when used to treat PDP is typically much lower (6.25–50 mg/day) versus the target dose used to treat conditions such as schizophrenia (300–800 mg/day) [56].
Recent International Parkinson and Movement Disorder Society (IPMDS) guidelines concluded that, with specialized monitoring, clozapine can be ‘clinically useful’ in the treatment of PDP because of its proven efficacy and acceptable risk [33]. These guidelines suggest that clozapine should be used in all patients with PDP who previously had treatment failure with quetiapine or pimavanserin. Furthermore, it may also be considered as a first-line option after tapering/discontinuing antiparkinsonian medication, despite onerous weekly blood count monitoring due to the risk of neutropenia. FDA guidance mandates weekly blood tests for patients receiving clozapine for the first 6 months along with absolute neutrophil count testing every 2 weeks for the following 6 months if an acceptable neutrophil count is maintained for the first 6 months. Absolute neutrophil count testing is required every 4 weeks thereafter [57]. Recent AGS Beers criteria include clozapine as an exception to the general recommendation to avoid antipsychotics in older patients with PDP because of its proven efficacy and minimal likelihood of worsening PD motor symptoms [41].
3.4 QuetiapineUnlike clozapine, most small clinical studies evaluating low-dose quetiapine in PDP have demonstrated little to no improvement in psychosis in PDP when compared with either placebo or clozapine [58,59,60,61]. However, quetiapine (25–50 mg) seems more tolerable when compared with clozapine, with lower risk of orthostatic hypotension (up to 7% with quetiapine compared with up to 19% with clozapine) and agranulocytosis (a recent report from a drug surveillance program of over 300,000 patients with psychotic symptoms treated with atypical antipsychotics showed a relative incidence of neutropenia and agranulocytosis of 0.23% with quetiapine vs. 1.57% with clozapine) [50, 51, 62, 63]. In other disease settings, quetiapine is associated with drowsiness (up to 57%) and weight gain (up to 23%) and may increase the risk of metabolic syndrome [53, 62]. Low-dose quetiapine (12.5–50 mg) also has limited effects on motor functioning in PDP, as seen in a recent systematic literature review that demonstrated that overall, quetiapine did not significantly worsen motor function versus placebo across several randomized trials, as measured by the UPDRS motor scores [64].
Recent IPMDS guidelines concluded that quetiapine can be ‘possibly useful’ in the treatment of PDP because of its acceptable safety profile, with no need for specialized monitoring despite the lack of high-quality evidence of efficacy in this setting [33]. They suggest that low-dose quetiapine can be considered a pragmatic first choice for PDP, if pimavanserin is unavailable, after the optimization of antiparkinsonian medication due to its improved safety profile compared with clozapine. Recent AGS Beers criteria also include quetiapine as an exception to the general recommendation to avoid antipsychotics in elderly patients with PDP, although they note quetiapine has only been evaluated in low-quality studies with comparable efficacy to both placebo and clozapine [41].
3.5 PimavanserinPimavanserin is a selective serotonin 5-hydroxytryptamine2A (5-HT2A) receptor inverse agonist and antagonist. Critically, pimavanserin also has no dopamine D2 receptor affinity [65] allowing it to, theoretically, be administered without altering the efficacy of dopaminergic agents that treat the motor systems associated with PD. In 2016, the FDA granted approval of pimavanserin for the treatment of H&D associated with PDP [66] based on the data from the ACP-103-020 (NCT01174004) trial.
3.5.1 Clinical DataPimavanserin was initially evaluated in two randomized placebo-controlled studies. The first study (ACP-103-006, NCT00087542) was a phase II, multicenter trial in 60 patients randomized 1:1 to receive 20–60 mg/day of pimavanserin versus placebo for a total of 4 weeks [67]. The key efficacy endpoint was improvement in the Scale for the Assessment of Positive Symptoms (SAPS). That was followed by two phase III trials evaluating pimavanserin versus placebo in PDP. The first, ACP-103-012 (NCT00477672), was a multicenter study in 259 patients with PDP randomized 1:1 to receive 10 or 40 mg/day of pimavanserin versus placebo for a total of 6 weeks [68]. The second, ACP-103-014 (NCT00658567) was a multicenter study in 123 patients with PDP randomized 1:1:1 to receive 10 or 20 mg/day of pimavanserin versus placebo for a total of 6 weeks [69]. The key efficacy endpoint for both phase III trials was improvement in the H&D component of the SAPS. While all three trials demonstrated the manageable safety profile of pimavanserin, including no worsening of parkinsonism as determined by the Unified Parkinson's Disease Rating Scale (UPDRS), neither trial showed a significant efficacy benefit over placebo in either the SAPS or SAPS H&D measurements, respectively. After completion of the phase III ACP-103-012 study, there were some concerns about the non-centralized assessment of psychotic symptoms globally versus in the US, in which there did appear to be a benefit with pimavanserin [68]. This led to the initiation of a subsequent North American-restricted phase III trial, ACP-103-020 (NCT01174004).
The ACP-103-020 trial was a phase III, randomized, double-blind, placebo-controlled study evaluating the use of pimavanserin for PDP [70]. After a 2-week lead-in phase of psychosocial therapy to induce a placebo response prior to baseline, 199 patients were randomized (1:1) to receive once-daily 40 mg pimavanserin tartrate, equivalent to 34 mg pimavanserin free base (n = 105), or placebo (n = 94) for up to 6 weeks. No reductions in dopaminergic drugs were required during the study. The primary outcome was the antipsychotic benefit as determined by the newly developed PD-adapted scale for assessment of positive symptoms (SAPS-PD). Key secondary outcomes included improvements in the Clinical Global Impression-Improvement (CGI-I) scores as well as safety and tolerability.
In the ACP-103-020 trial, pimavanserin was reportedly associated with a 5.79-point decrease in SAPS-PD compared with a 2.73-point decrease with placebo (Δ − 3.06 points; 95% CI − 4.91 to − 1.2; p = 0.001) [70]. At the end of the 6-week period, 73.7% of patients who received pimavanserin achieved at least a 1-point improvement (i.e., decrease) in SAPS-PD from baseline, 33.7% achieved an improvement of at least 10 points, and 13.7% achieved a complete response, compared with 55.6%, 16.7%, and 1.1% with placebo, respectively [66]. Similarly, pimavanserin significantly improved CGI-I compared with placebo (2.78 vs. 3.45; p = 0.0011). The caregivers of patients receiving pimavanserin reported a larger reduction in burden-of-care than caregivers of patients in the placebo arm (4.34-point improvement in the Zarit 22-item caregiver burden scale; 95% CI − 7.00 to −1.67; p = 0.0016). These, along with other measurements, demonstrated that pimavanserin is efficacious when compared with placebo in the treatment of PDP [70].
The incidence of treatment-emergent adverse events in the ACP-103-020 trial was similar between the pimavanserin and placebo arms, with the most common being urinary tract infections (UTIs; 13% vs. 12%), falls (11% vs. 9%), hallucinations (7% vs. 4%), peripheral edema (7% vs. 3%), nausea (6% vs. 6%), and confused state (6% vs. 3%). Eleven percent of patients in the pimavanserin group, versus 4% in the placebo group, experienced serious adverse events. A mean increase of 7.3 ms in QT interval from baseline to 43 days was observed in the pimavanserin arm versus no change in the placebo arm. Unlike other noted atypical antipsychotics (e.g., clozapine and quetiapine), there was no association of pimavanserin with weight gain, somnolence, or metabolic syndrome. Notably, there was no evidence of treatment-related impairment of motor function in either the pimavanserin or placebo arms [70].
A more recent, open-label extension study (NCT00550238) demonstrated similar results [71, 72]. At the end of a 4-week extension period, the mean change in SAPS-PD at 10 weeks from baseline for all patients was − 1.8 (95% CI − 2.3 to − 1.2), and the subgroup of patients who had not received prior pimavanserin experienced a mean SAPS-PD benefit of − 2.9 (95% CI − 3.8 to − 2.1) [72]. 68.0% of patients in the extension study continued pimavanserin for 6 months and 18.1% continued pimavanserin for more than 4 years [71]. After prolonged exposure to pimavanserin, the adverse event profile resembled that observed in the placebo-controlled 6-week pimavanserin studies and most adverse events were mild or moderate. Additionally, early response to pimavanserin seemed to be durable with extended treatment.
3.5.2 Evaluating Mortality Risk with PimavanserinRecently, three insurance database retrospective analyses evaluated the risk of mortality associated with pimavanserin versus other atypical antipsychotics. One study using commercially insured patients receiving atypical antipsychotics to treat PDP found that there was no significant difference in the risk of mortality with pimavanserin versus the preferred atypical antipsychotics quetiapine and clozapine (adjusted hazard ratio [HR] 0.99, 95% CI 0.81–1.20) or non-preferred atypical antipsychotics (adjusted HR 0.98, 95% CI 0.79–1.22) [73]. A second study assessing Medicare beneficiaries with PD showed that the risk of mortality with pimavanserin was approximately 35% lower than with other atypical antipsychotics (quetiapine, risperidone, olanzapine, or aripiprazole) within the first 180 days of treatment (HR 0.65, 95% CI 0.53–0.79), but there was no significant difference in mortality with treatment beyond 180 days (HR 1.05, 95% CI 0.82–1.33) [74]. Another study assessing Medicare beneficiaries with PD showed a similar improvement in risk of mortality with pimavanserin versus other atypical antipsychotics (clozapine, quetiapine, risperidone, olanzapine, aripiprazole, or brexpiprazole) with an HR of 0.78 (95% CI 0.67–0.91) [
留言 (0)