
記住我
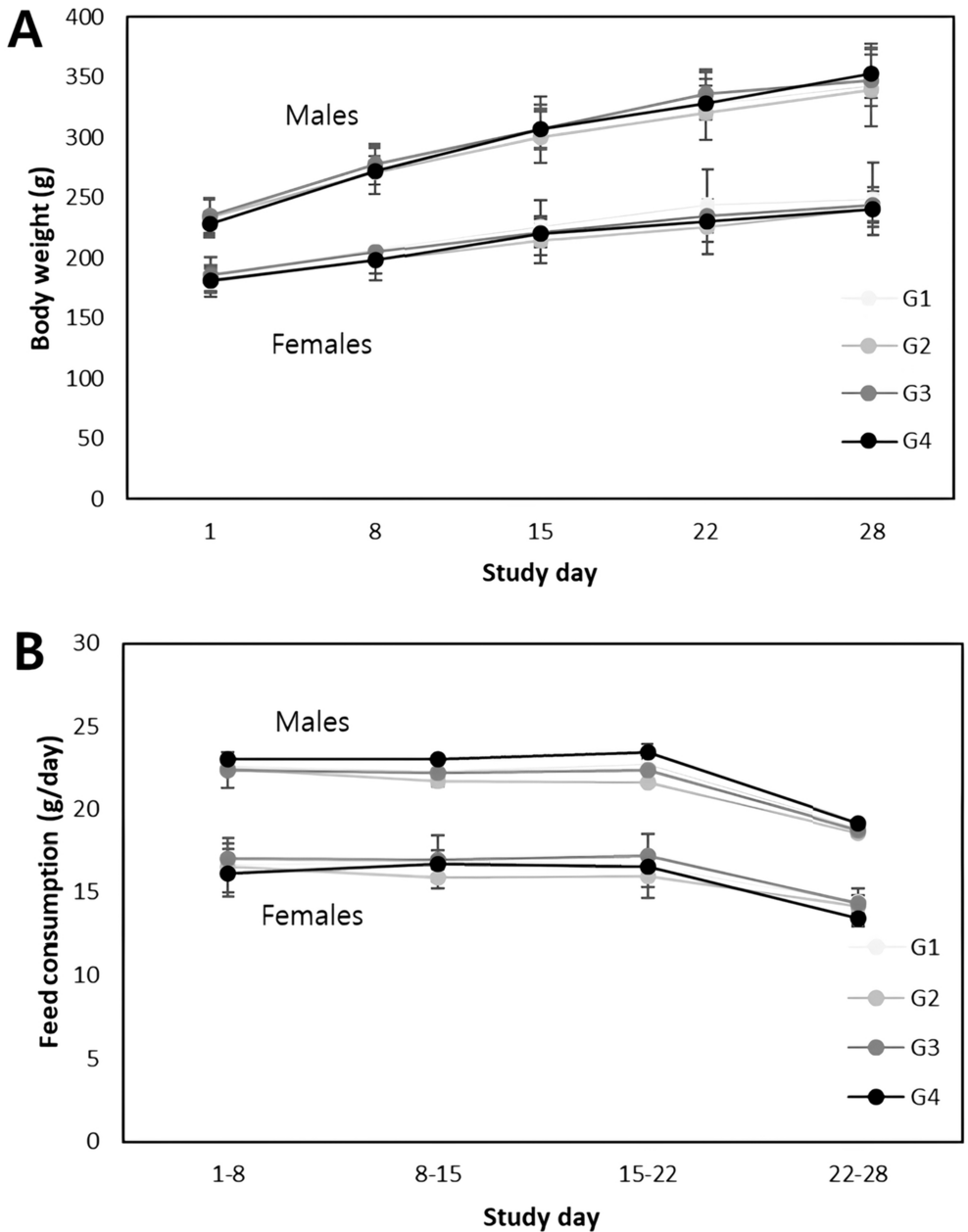


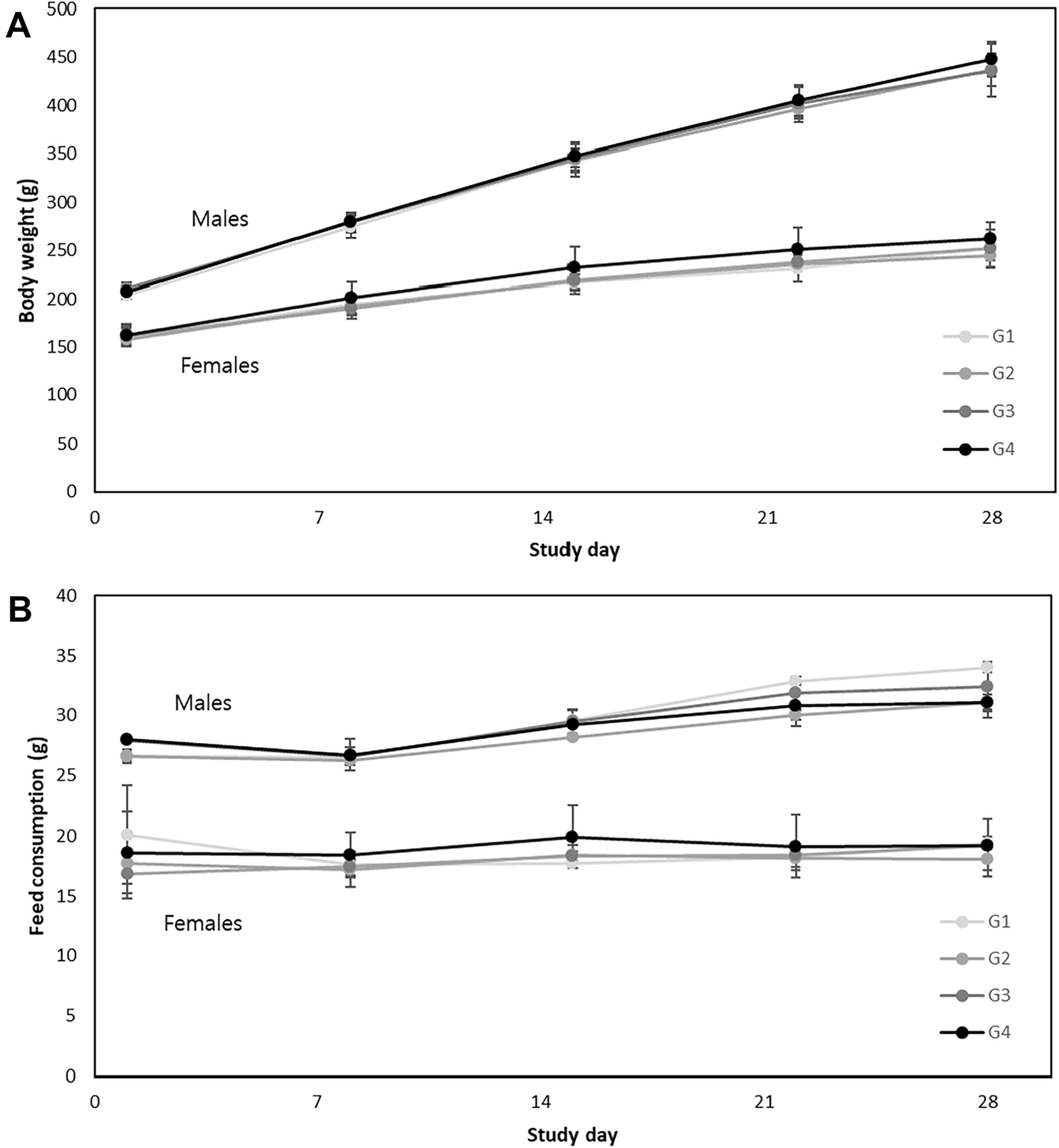

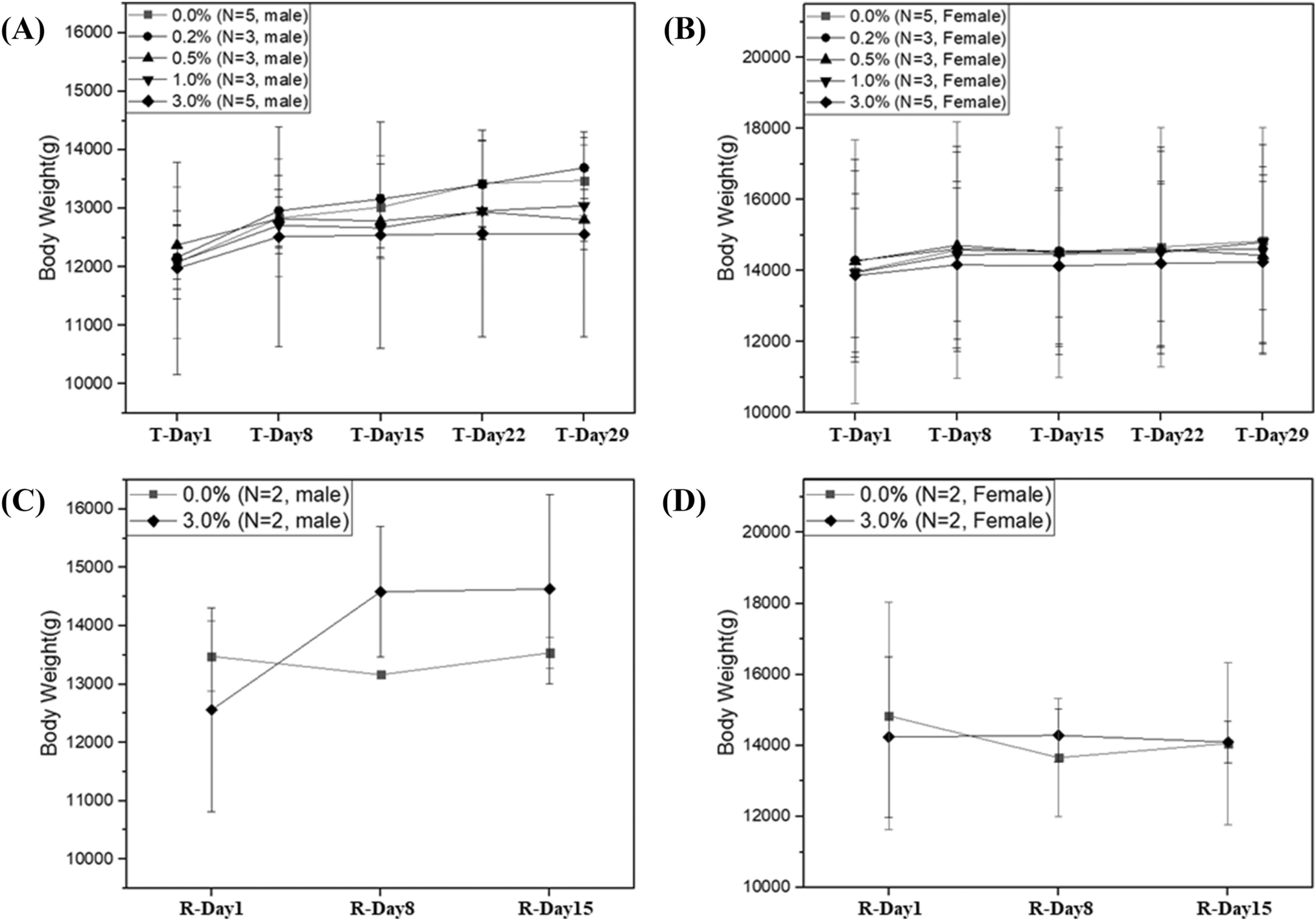




All reagents used were of analytical grade. Ethanol, ethylenediaminetetracetic acid disodium (Na2EDTA) and tetrasodium (Na4EDTA) salt, sodium chloride (NaCl), and sodium hydroxide (NaOH) were purchased from Carlo Erba Reagenti Srl (Milan, Italy). Dimethyl sulfoxide (DMSO), ethidium bromide, hydrocortisone hemisuccinate, insulin, low- and normal melting-point agarose (LMPA and NMPA, respectively), 4-nitroquinoline N-oxide (4NQO), staurosporine, tris(hydroxymethyl)aminomethane (Tris base), Triton X-100, trypan blue, and valinomycin were obtained from Sigma-Aldrich Srl (Milan, Italy). Acridine orange (AO), 6,4′-diamidino-2-phenylindole (DAPI), 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolecarbocyanine iodide (JC-1), Via1-Cassette™, and NC-Slide A8™ were purchased from ChemoMetec A/S (Allerød, Denmark). Eagle’s Minimum Essential Medium (MEM), and Dulbecco’s phosphate-buffered saline, pH 7.4 (PBS) were purchased from Invitrogen Srl (Milan, Italy). Countess™ cell counting chamber slides, Enhanced chemiluminesce (ECL) detection kit, Gibco™ William’s E medium, Glutamax and Pierce™ IP lysis buffer were from Thermo Fisher Scientific (Waltham, MA, USA). Antibiotics (penicillin and streptomycin), foetal bovine serum (FBS), L-glutamine, MEM non-essential amino acids (NEAA), sodium pyruvate, and trypsin were purchased from Euroclone SpA (Milan, Italy). Anti-Caspase-3 (E-AB-60017) and anti-β-Actin (E-AB-20031) antibodies were purchased from Elabscience (Houston, TX, USA). Anti-mouse and anti-rabbit horseradish peroxidase (HRP)-linked secondary antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). Conventional microscope slides and coverslips were supplied by Knittel-Glaser GmbH (Braunschweig, Germany). Distilled water was used throughout the experiments.
Phenylselenenylzinc chloride (PhSeZnCl)PhSeZnCl (Fig. 1) was synthesised in the laboratory of Professor Claudio Santi as previously described [16] by refluxing equimolar amounts of PhSeCl and freshly activated zinc in dry THF. The top concentration used was the highest soluble concentration obtained—as previously described [22]—according to the US National Toxicology Program Interagency Center for the Evaluation of Alternative Toxicological Methods (NICEATM) [23] and abiding by the US Food and Drug Administration [24] and OECD guidelines for testing pharmaceuticals intended for human use [25].
Fig. 1
Briefly, PhSeZnCl dry powder was firstly suspended in DMSO until the concentration of 50 mg/mL, and then further diluted in complete medium until desired concentrations, namely 500 µg/mL.
Cell lines and culture conditionsThis research aimed to evaluate the cytotoxic, genotoxic, and apoptotic activities, as well as effects on cell cycle of PhSeZnCl in two preclinical hepatic models, namely HepG2 and HepaRG cells.
HepG2 cells originate from a clone of human tumour cells isolated in 1975 from the liver of a young Argentine of 15 years with a diagnosis of hepatoblastoma [26,27,28]. These cells are commonly used for toxicity investigation due to their unlimited life span, stable phenotype, high availability, and easy handling [29]. Moreover, it has been shown that the use of such an in vitro model is more adherent to the situation in vivo, compared with the addition of exogenous metabolic activation systems (e.g., S9-mix) in the case of use of other lines with lower metabolic capabilities [30].
The human HepG2 cell line (ATCC HB 8065) was obtained from Istituto Zooprofilattico Sperimentale della Lombardia e dell’Emilia Romagna “Bruno Ubertini” (Brescia, Italy). The cells were grown as monolayer cultures in 25 cm2 tissue flasks in MEM supplemented with 10% (v/v) FBS, 1% NEAA, 1 mM sodium pyruvate, 100 U/mL penicillin and 0.1 mg/mL streptomycin, at 37 °C in a humidified atmosphere containing 5% CO2. HepG2 cells were sub-cultured by dispersal with 0.05% trypsin in 0.02% Na4EDTA for a contact time of 5 min and replated at a 1:2 dilution to maintain the cells in the exponential growth phase. For cell treatment, sub-confluent HepG2 cultures were collected by trypsin treatment and suspended in complete MEM culture medium.
The experimental design also included tests on undifferentiated HepaRG™ liver cells. HepaRG cells derive from a tumour of a female patient suffering from chronic hepatitis C infection and hepatocellular carcinoma [31]. Regardless of their differentiation status, HepaRG cells are closer to primary human hepatocytes and liver tissues compared with HepG2 cells [32]. For these reasons, HepaRG cells are considered to be a useful model for in vitro studies on drug metabolism and toxicity [33, 34].
The human HepaRG hepatic cells were purchased by Thermo Fisher Scientific (Waltham, MA, USA). The cells were thawed, seeded at low density (2 × 104 cells/cm2) in 75 cm2 flasks and maintained in William's E medium supplemented with 1% Glutamax, 5 μg/mL human insulin, 50 μM hydrocortisone hemisuccinate, 100 U/mL penicillin, 0.1 mg/mL streptomycin and 10% FBS at 37 °C in a humidified atmosphere containing 5% CO2. When high confluence was achieved (at day 14), cells were sub-cultured, in order to maintain the cell line. Medium was renewed every 2 or 3 days.
Cytotoxicity testing: trypan blue dye exclusion assayWe assessed the possible cytotoxicity of PhSeZnCl by evaluating 6 scalar concentrations (i.e., 6.25, 12.5, 25, 50, 100, and 500 µg/mL); the top concentration was based on the US Food and Drug Administration guidelines for testing pharmaceuticals intended for human use [24]. The trypan blue dye exclusion assay is based on the principle that live cells possess intact cell membranes that exclude certain dyes, such as trypan blue, whereas dead cells do not. The reactivity of trypan blue is based on the fact that the chromophore is negatively charged and does not interact with the cell unless the membrane is damaged; therefore, the cells that exclude the dye are considered viable [35].
For the test, cells (5 × 105 per well) were dispensed within six-well culture plates (Becton Dickinson Italia SpA, Milan, Italy) in 5 mL volumes. Cells were maintained in culture for 24 h to form a semiconfluent monolayer and then treated with 2% Triton X-100 (positive control) or PhSeZnCl over a range of six concentrations. The cells were exposed for 4 or 24 h. Cytotoxicity was measured using a Countess™ (Invitrogen Srl, Milan, Italy) automated cell counter [36]. Briefly, aliquots of cell suspensions were mixed with equal volumes of 0.4% trypan blue and 10 µL loaded onto a Countess cell counting chamber slide. The Countess counter is equipped with a camera that acquires images from cell samples on the chamber slide, the image analysis software automatically analyses the acquired cell images and measures cell count and viability.
Cytotoxicity testing: AO/DAPI double stainingThe number of total and viable cells was also estimated by staining cell populations with AO and DAPI fluorophores. For the test, the cells were cultured as above described for the trypan blue exclusion assay. After cell treatment, aliquots of cell suspensions were loaded into Via1-Cassette. The inside of the Via1-Cassette is coated with AO (staining the entire population of cells) and DAPI (staining nonviable cells). The Via1-Cassettes were then placed in a NucleoCounter® NC-3000™ (Chemometec, Allerød, Denmark), a fluorescence-based image cytometer, where cell concentration and viability were determined [36, 37]. Total cell concentration in Via1-Cassette was displayed on a personal computer using the NucleoView software.
Genotoxicity testing: comet assayTo avoid conditions that would lead to false-positive results arising from DNA damage associated with cytotoxicity [38], the three highest non-cytotoxic concentrations of PhSeZnCl (i.e., 12.5, 25, and 50 µg/mL) were processed in the comet assay [39]. The test was conducted under alkaline conditions (alkaline unwinding/alkaline electrophoresis, pH > 13) following the original three-layer procedure [39] as described in detail elsewhere [36].
Briefly, 48 h prior to PhSeZnCl treatment, HepG2 and HepaRG subcultures were trypsinized and seeded (approximately 5 × 105 cells/well) in six-well plates. Cells were then treated for 4 h with PhSeZnCl; cell subcultures were also treated with the model mutagen 4NQO (1 µM; positive control). One hundred cells were randomly selected and analysed from each experimental point. The percentage of DNA in the comet tail (i.e., tail intensity %) was used as a measure of the extent of DNA damage [40]. The percentage of hedgehog cells (cells with tail intensity of around 85–90% and above) were reported and hedgehog comets were included in the analysis of genotoxicity [41].
Analysis of cell cycleFor cell cycle analysis, HepG2 and HepaRG cells were cultured overnight in six-well plates at 5 × 105 cells per well. Cell cultures were then exposed to PhSeZnCl (i.e., 12.5, 25, and 50 µg/mL) for 24 h. At the end of the treatment, the cells were harvested, fixed with 70% ethanol, and maintained at 0–4 °C for at least 24 h. After centrifugation, cell pellets were washed with PBS and treated for 5 min at 37 °C with 0.5 mL of PBS containing 1 µg/mL DAPI and 0.1% Triton X-100. After staining, DAPI fluorescence was quantified by fluorescence microscopy with the automated cytometer NucleoCounter NC-3000 [42]. The stained cells were analysed using the NucleoView NC-3000 software and obtained results were presented in the DNA content histograms where different phases of the cell cycle were demarcated.
Early apoptosis: evaluation of mitochondrial membrane potential (ΔΨm) assayThe cells were cultured as above and exposed to PhSeZnCl for 4 and 24 h. Valinomycin 0.5 µM was used as a positive control. Following treatment, mitochondrial membrane potential (ΔΨm) was estimated by a NucleoCounter NC-3000 automated system after staining of cells with JC-1 and DAPI [43]. In early apoptotic cells, where the mitochondrial membrane potential collapses, the monomeric JC-1 remains cytosolic and stains the cytosol with a green colour. On the other hand, in non-apoptotic cells, JC-1 rapidly forms complexes with intense red fluorescence [36, 44]. Mitochondrial membrane depolarization was revealed as a decrease in the red/green fluorescence intensity ratio. The scatterplots obtained by the NucleoView NC-3000 software were used to demarcate the percentage of polarized/healthy cells, depolarized/early apoptotic cells and DAPI positive/late apoptotic or necrotic cells, respectively [45].
Late apoptosis: chromosomal DNA fragmentation assayHepG2 and HepaRG cells were cultured as above described for cell cycle analysis and exposed to PhSeZnCl for 4 and 24 h. Staurosporine (1 μM) was used as a positive control. Fragmentation of chromosomal DNA during late apoptosis was revealed by discrete sub-G1 peaks on DNA content histograms [46]. At the end of the treatment, a NucleoCounter NC-3000 automated system using fluorescence microscopy and image analysis was used to determine the extent of DNA fragmentation [45]. For the test, the cells were first permeabilized with ethanol; during this procedure, the low molecular weight DNA inside apoptotic cells leaks out and is removed from the sample during the subsequent washing step. The high molecular weight DNA retained in the cells was stained with DAPI.
Western immunoblot analysisActivation of Caspase-3—an effector in most of apoptotic pathways—was also investigated by Western Blot analysis. HepG2 and HepaRG cells were seeded in T25 flasks at 2 × 106 cells/flask and cultured overnight. Cells were exposed to PhSeZnCl (i.e., 12.5, 25, and 50 µg/mL) for 4 and 24 h. Staurosporine (1 μg/mL) was used as positive control. After treatment, cells were scraped and lysed with ice-cold Pierce IP lysis buffer. Lysates were centrifuged at 12,000×g and supernatant containing cell proteins was collected. The protein amount in each lysate was determined by Bradford’s method. Sixty micrograms of total proteins for each sample were resolved on a 16% SDS-Page gel and blotted on a nitrocellulose membrane (0.2 μm pore). Membranes were blocked for 1 h at room temperature with 5% skim milk in PBS containing 0.05% Tween-20. For immunodetection, nitrocellulose membranes were incubated overnight with 1:1000 anti-Caspase-3 antibody at 4 °C. β-Actin was used as loading control. Then, membranes were washed in T-PBS and incubated in anti-mouse (for β-Actin) or anti-rabbit (for Caspase-3) (HRP)-linked secondary antibody for 2 h at room temperature. Protein signals were detected using ECL detection kit (Thermo Fisher Scientific) and the ImageQuant LAS 500 (GE Healthcare, Milan, Italy). Densitometric analysis was carried out using ImageJ software (https://imagej.nih.gov/ij/).
Statistical analysisThe assays were carried out at least in triplicate. After testing the normal distribution of data with the Kolmogorov–Smirnov test, the results were expressed as the mean ± standard error of the mean (SEM). Differences were investigated by one-way analysis of variance (ANOVA) followed by Dunnett’s post hoc analysis for pairwise comparisons between PhSeZnCl and untreated (control) cells. Student’s t-test was used for positive controls. The level of significance was set at p < 0.05. IC50 was estimated by considering logarithm transformed PhSeZnCl concentrations.
留言 (0)