Test substance and vehicle
The test substance was W. cibaria CMU (oraCMU®, OraPharm, Inc., Seoul, Korea) in a vehicle of distilled water (Daihan Pharma Co., Seoul, Korea). The W. cibaria preparation contained 3.6 × 108 CFU/g. Preparations used for each study were prepared daily. For the in vivo studies, samples were collected from the dosage formulations to verify homogeneity and dose concentration on the day of administration.
Cell viability was determined using an EZ-Cytox cell viability assay kit (iTSBiO, Korea). Briefly, EZ-Cytox kit reagent was added into each well for 2 h under standard culture conditions, after which 96-well plates were gently shaken thoroughly for 5 min on a rocker at room temperature. The absorbance of the treated and untreated samples at 450 nm was then measured on a multi-well microplate reader (VersaMax, Molecular devices, San Jose, CA, USA). DMEM medium supplemented with the same volume of kit reagent on an empty well was used as a blank. Cell viability was represented by percentage values compared to a control and then used to calculate the number of viable cells.
Study conduct
All studies conducted in animals complied with the guidelines of animal ethics and were approved by the IACUC of Korea Conformity Laboratories (KCL). Approval numbers for each study are as follows: In vivo micronucleus test (IA20-02198); single oral dose toxicity study (IA20-01118); thirteen-week repeated oral Dose toxicity study (IA20-01554). All studies were performed at KCL in Incheon, Korea.
The 14-day dose range finding and 13-week studies were performed according to OECD guidelines 407 and 408, respectively [12, 13]. OECD guidelines 417, 473 and 474 were followed for the bacterial reverse mutation study, the in vitro chromosomal aberration test, and the in vivo micronucleus test, respectively [14,15,16]. All studies were conducted in compliance with non-clinical trial management standards as stated in the Korean Ministry of Food and Drug Safety (MFDS) Notification No. 2018-93, “Principles of Good Laboratory Practices (GLP)” (November 21, 2018), except for the 14-day oral dose range finding study. All of the studies were also performed according to MFDS Notification No. 2017-71, “Toxicity Test Standards of Medicine and Medicinal Supplies” (August 30, 2017).
Animals
Specific Pathogen Free (SPF) Sprague–Dawley (SD) rats from Orient Bio Inc. (Seongnam, Korea) were used in this study. Animals were acclimated for 5–8 days (depending on the study) before treatment and were approximately 8 weeks of age at the start of the acute study, and 6 weeks for the 14-day and 13-week studies. Animals were housed in wire mesh cages in groups no larger than three for the acute and 14-day studies and five for the 13-week study. The animal housing room was maintained on a 12-h light/dark cycle, with temperature and relative humidity of 24 ± 0.3 °C and 46.3 ± 1.5% in the acute study, 23.3 ± 0.5 °C and 51.1 ± 0.5% in the 14-day study and 22.7 ± 0.4 °C and 54.9 ± 1.5% in the 13-week study. Animals were supplied diet (Teklad Certified Irradiated Global 18% Protein Rodent Diet, Envigo, USA) and filtered water ad libitum. Animals were selected for use in the study only if their body weight fell within 20% of the mean. For the acute study, animals were fasted overnight prior to dosing and food was returned three to four hours after dosing; water was provided ad libitum during fasting. For the 14-day and 13-week studies, rats were randomly allocated into treatment groups to ensure adequate distribution of body weights.
Single dose oral toxicity study
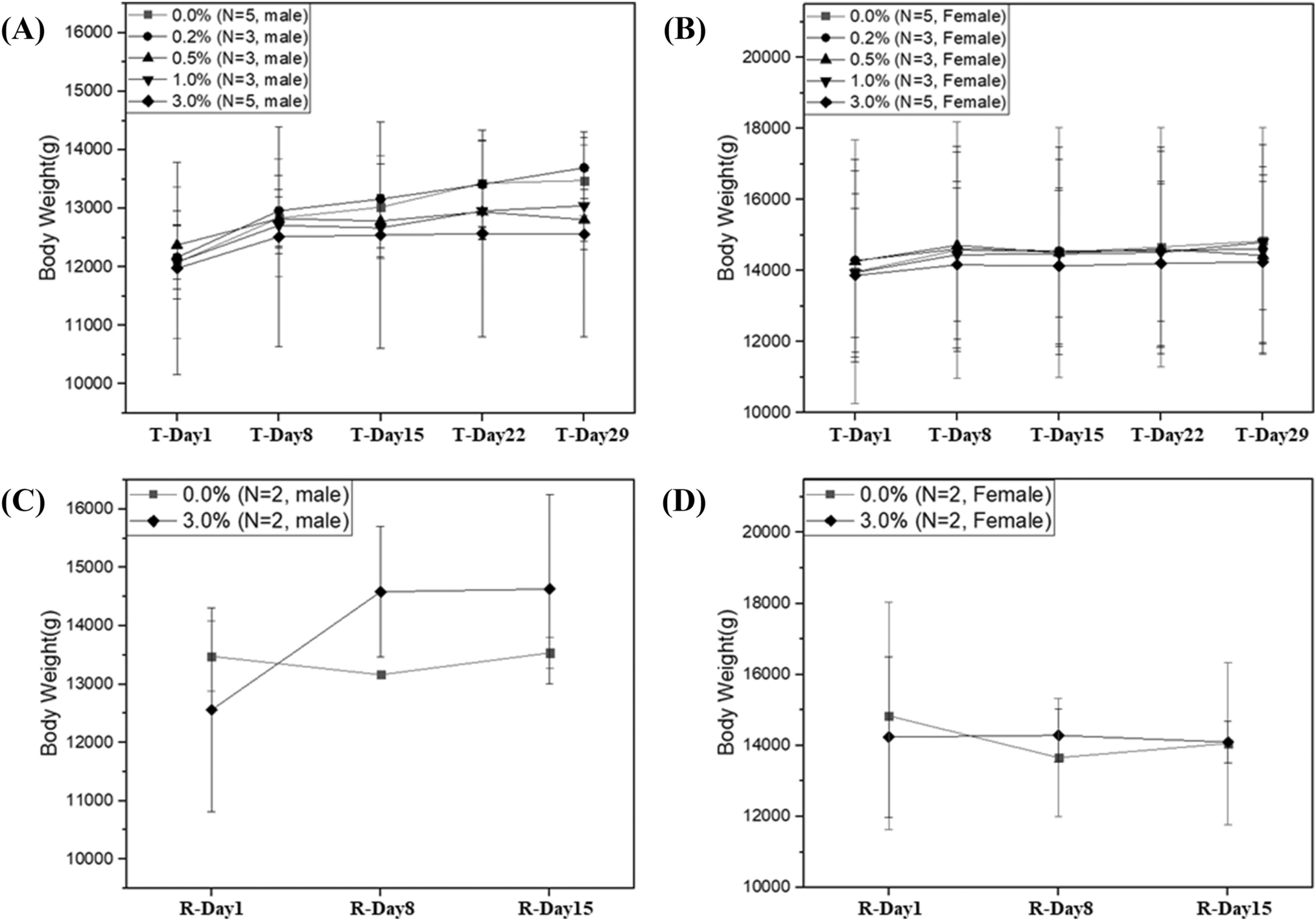
Animals were divided into four groups (n = 5/sex/group), one of which was a vehicle control group (distilled water), and three test groups receiving the test material at 1250, 2500, or 5000 mg/kg body weight (bw). The viable cell counts in the low, medium, and high dose groups were 4.5 × 108, 9.0 × 108, and 1.8 × 109 CFU/kg, respectively. The test material was prepared just prior to dosing by dissolving the test material in distilled water at the appropriate concentration. Animals were dosed by gavage at a volume of 15 mL/kg bw. Following dosing, general clinical signs in all animals were observed more than once a day for 14 days before necropsy. On the day of administration, clinical signs were observed in all animals at 30 min and every hour until 6 h after administration. Body weights were taken at acquisition, grouping, immediately prior to dosing, and on days 1, 4, 7, and 14 after dosing. On day 14, animals were anesthetized with carbon dioxide (CO2) and humanely euthanized. Complete postmortem examinations were performed on all organs.
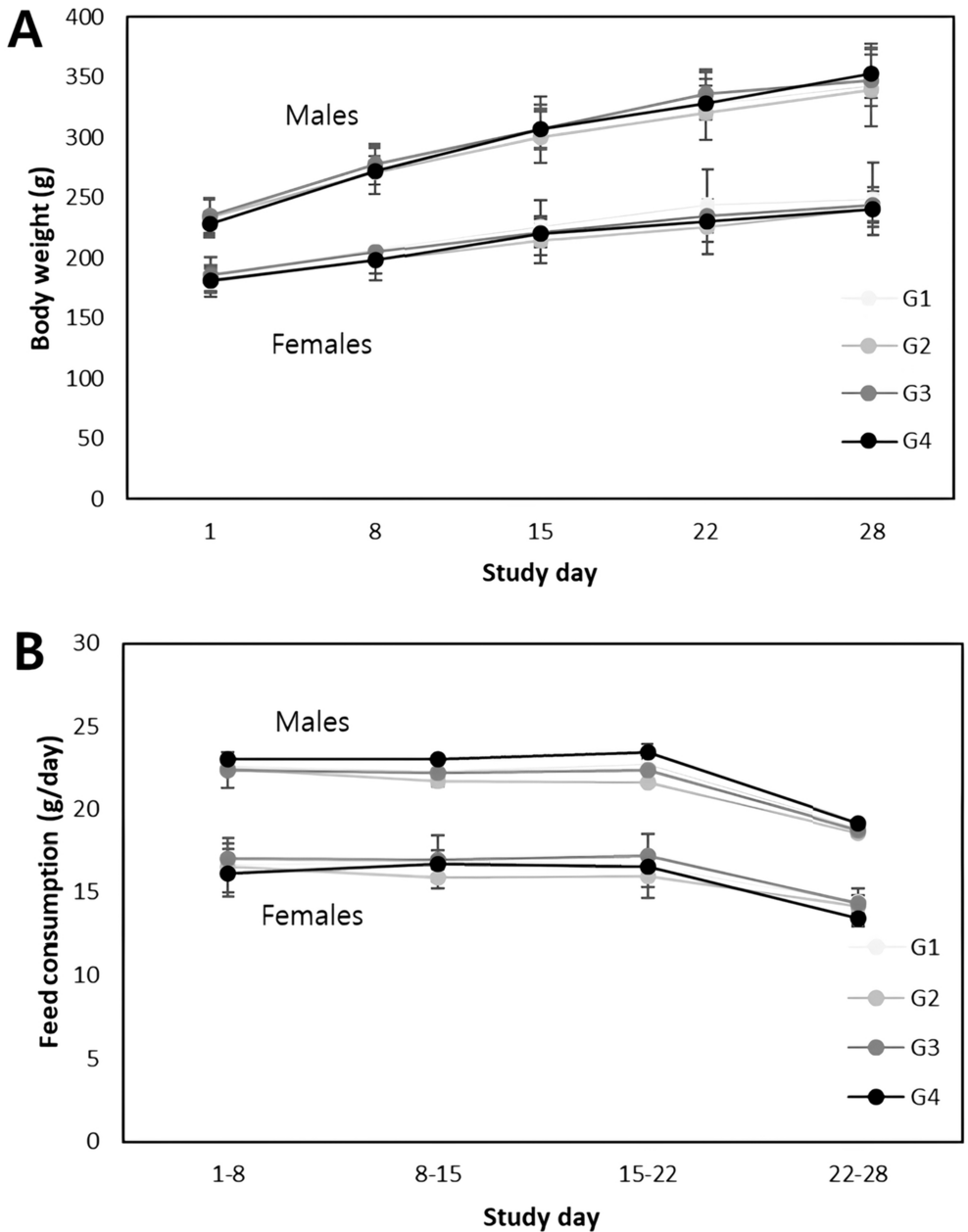
14-day dose range finding study
In total, 40 rats (n = 20/sex) were assigned to four treatment groups (n = 5/sex/group)—vehicle control (Group 1) or the following amounts of the test substance: 4.5 × 108 CFU/kg bw/day (Group 2), 9.0 × 108 CFU/kg bw/day (Group 3) or 1.8 × 109 CFU/kg bw/day (Group 4) for 14 days. Individual doses were calculated based on the most recent weekly body weights and were adjusted each week to maintain the targeted dose level for all rats (i.e., mg/kg bw/day). All doses were administered volumetrically at 15 mL/kg by oral gavage.
General clinical signs of all animals were observed and recorded daily. Body weights were recorded at acquisition, grouping, before administration, once a week during the study, and before necropsy. Food intake was measured right before the first administration and once a week during the study. Food consumption per animal was measured by the average intake (g/rat/day) of individual animals in a cage. Water consumption was measured in a similar manner to food consumption. The night before termination, the animals were subjected to an overnight fast in which water was still provided ad libitum. At study termination, animals were euthanized under isoflurane anesthesia and blood samples were drawn from abdominal aorta. Whole blood collected on EDTA-K2 was used for hematological analyses (white blood cell count (WBC), neutrophils (NEU), eosinophils (EOS), basophils (BAS), lymphocytes (LYM), monocytes (MON), large unstained cells (LUC), percent of neutrophils (NEP), percent of eosinophils (EOP), percent of basophils (BAP), percent of lymphocytes (LYP), percent of monocytes (MOP), percent of large unstained cells (LUP), red blood cell count (RBC), hemoglobin concentration (HGB), red cell distribution width (RDW), hematocrit (HCT), mean corpuscular volume (MCV), mean corpuscular hemoglobin (MCH), mean corpuscular hemoglobin concentration (MCHC), reticulocytes (RET), platelets (PLT), and mean platelet volume (MPV)). Serum was obtained from whole blood collected on 3.2% sodium citrate and analyzed for clinical chemistry (aspartate aminotransferase (AST), alanine aminotransferase (ALT), gamma-glutamyl transferase (GGT), alkaline phosphatase (ALP), total bilirubin (BIL), blood urea nitrogen (BUN), creatinine (CRE), uric acid (UA), glucose (GLU), total cholesterol (CHO), triglycerides (TG), total protein (PRO), albumin (ALB), ALB/GLOB ratio, lactate dehydrogenase (LDH), creatine phosphokinase (CPK), calcium (Ca), inorganic phosphorus (IP), magnesium (Mg), sodium (Na), potassium (K), and chloride (Cl)).
At necropsy, postmortem examinations were completed on all organs. Weights of the following organs were obtained: testes, prostate, ovaries, uterus, spleen, liver, thymus, adrenals, kidneys, heart, lungs, brain, and pituitary gland. All collected organs were preserved in 10% phosphate-buffered formalin except for testes, which were fixed in Böuin solution. Histopathological examination was not performed.
13-week repeated dose toxicity study
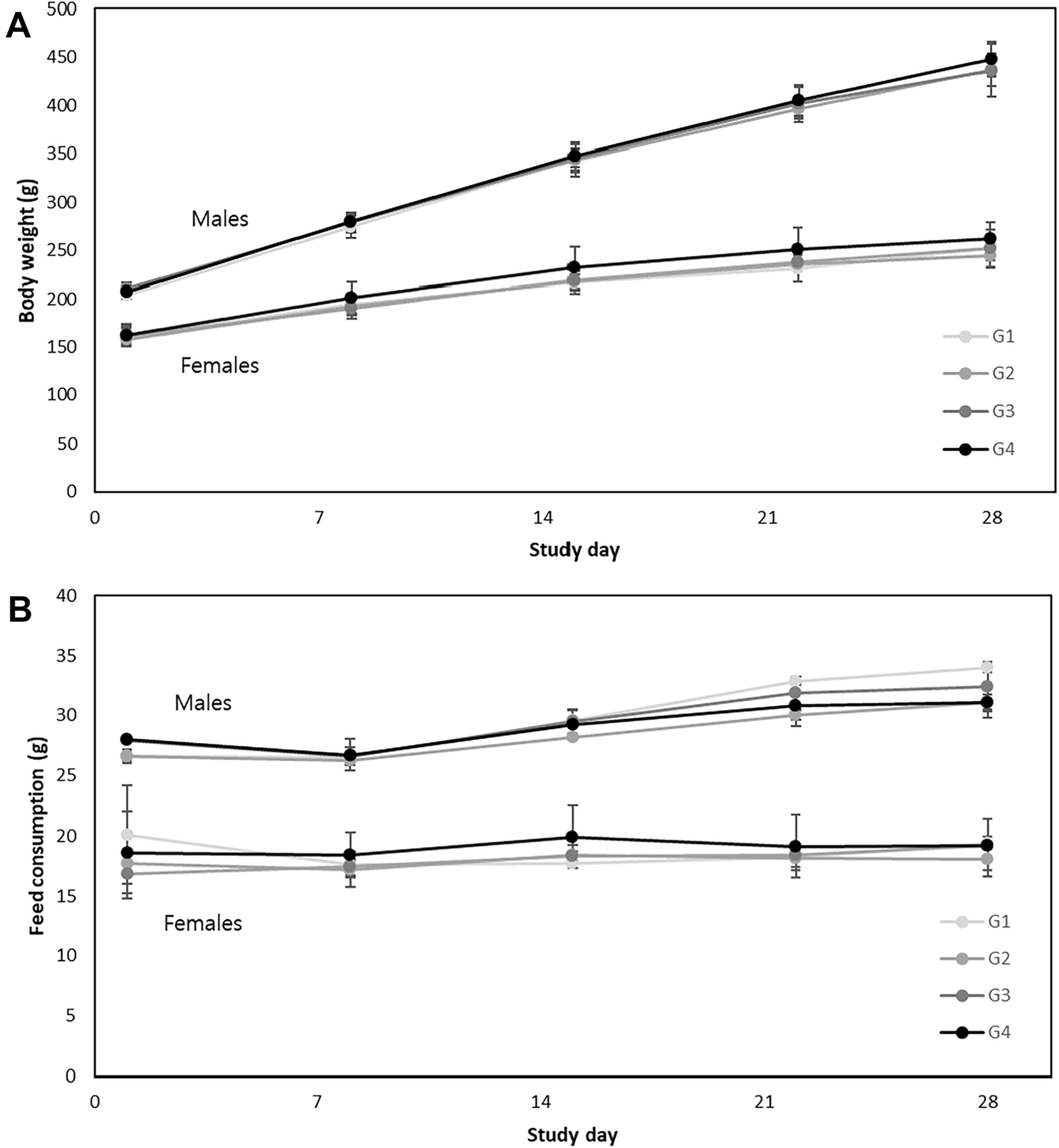
Eighty animals (n = 40/sex) were assigned to four treatment groups (n = 10/sex/group)—vehicle control (Group 1), and three treatment groups receiving the following amounts of test substance: 4.5 × 108 CFU/kg bw/day (Group 2), 9.0 × 108 CFU/kg bw/day (Group 3) or 1.8 × 109 CFU/kg bw/day (Group 4). Dose levels were adjusted based on body weights measured right before each administration, dosing volume was 15 mL/kg bw/day, and each dose was given by oral gavage for 13 weeks.
General clinical signs were observed and recorded daily for all animals. Individual body weights were recorded at acquisition, grouping, right before administration, once a week during exposure, and right before necropsy. Food consumption was measured right before first administration and once a week during the rest of the study. Food intake per day and food intake per animal were calculated the same way as for the 14-day dose range finding study. Ophthalmic examinations were conducted on all animals before first administration and during the last week of the study. During the last week of test substance administration, urine was collected from five animals per sex per group using metabolic cages. Urine samples were analyzed for the following parameters: color, glucose, bilirubin, ketone bodies, specific gravity, occult blood, pH, protein, urobilinogen, nitrite, leukocytes, erythrocytes, epithelial cells, and casts. Animals were fasted overnight before termination, but water was provided ad libitum. On the day of termination, all animals were anesthetized with isoflurane and blood samples were drawn from the abdominal aorta. Blood samples collected on EDTA-K2 were analyzed for the same hematology parameters as described for the 14-day dose range finding study, but methemoglobin (metHGB) was added. Analyses for coagulation (prothrombin time (PT) and active partial thromboplastin time (APTT)) were added. Serum clinical chemistry was also the same as the 14-day study, with the addition of bile acid (BA), high density lipoprotein (HDL), low density lipoprotein (LDL), cholinesterase (ChE), thyroxine (T4), triiodothyronine (T3), and thyroid stimulating hormone (TSH).
Scheduled necropsy was completed on all surviving animals and complete postmortem examinations were performed on all organs. Vaginal smear examinations were completed in all female rats prior to necropsy to determine estrus cycle by the shape of epithelial cells using Giemsa staining (Sigma, St. Louis, MO, USA). Weights of the 13 organs weighed in the 14-day study, plus the epididymides and thyroid, were taken at necropsy. Both sides of bilateral organs were weighed and preserved, and the thyroid was fixed prior to weighing. Organs and tissues from all animals were preserved in 10% neutral phosphate-buffered formalin (other than the testes and epididymides, which were fixed in Böuin solution, and the eyes which were fixed in Davison’s solution). Preserved organs included the brain, pituitary, heart, lungs, liver, kidney, urinary bladder, mesenteric lymph node, thymus, spleen, adrenal gland, esophagus, aorta, spinal cord, sciatic nerve, skeletal muscle, skin, mammary gland, eye, stomach, colon, rectum, femur, sternum and bone marrow, trachea, tongue, prostate gland, testes, epididymis, seminal vesicle, pancreas, salivary gland, submandibular lymph node, thyroid and parathyroid glands, duodenum, jejunum, ileum, cecum, ovary, uterus, cervix, and vagina. Specimens from the vehicle control and high dose group were prepared and examined for all fixed organs and tissues.
Bacterial reverse mutation assay
Bacterial strains utilized in all experiments were Salmonella typhimurium tester strains TA98, TA100, TA1535 and TA1537 and Escherichia coli WP2uvrA. Experiments were conducted in the absence and presence of an S9 metabolizing system, using the preincubation method. The test product was formulated as a solution in distilled water to provide dose levels of up to 5000 μg/plate. The positive control chemicals sodium azide (NaN3), 9-aminoacridine (9-AA), 2-(2-furyl)-3-(5-nitro-2-furyl) acrylamide (AF-2), 2-aminoanthracene (2-AA), benzo(a)pyrene (BP), and 4-nitroquinoline 1-oxide (4-NQO) and their vehicle dimethyl sulfoxide (DMSO) were sourced from Sigma, with the exception of AF-2 and DMSO, which came from Wako (Tokyo, Japan) and Junsei (Tokyo, Japan) (respectively). The bacterial strains and the S9 tissue fraction (which was isolated from livers of SD rats induced by intraperitoneal injection of Aroclor-1254) were sourced from Molecular Toxicology, Inc (Boone, NC, USA).
For the experiments, the following substances were mixed in a test tube and poured over the surface of a minimal glucose agar plate (Molecular Toxicology, Inc.): (1) the positive or negative control solutions or test formulations (0.05 mL each); (2) 0.5 mL of 0.1 M phosphate buffered saline or 0.5 mL of the S9 mix (for metabolic activation); (3) 0.1 mL of bacterial suspension; and (4) 2.0 mL of overlay agar supplemented with biotin and limited amounts of histidine and tryptophan (Molecular Toxicology, Inc.). Concentrations of test article used were 62, 185, 556, 1667, and 5000 μg/plate. The positive controls in the absence of S9 mix were AF-2 for S. typhimurium TA98 and TA100, sodium azide for TA1535, 9-AA for TA1537 and 4-NQO for E. coli WP2uvrA. The positive control for all bacterial strains in the presence of S9 mix was 2-AA, except for TA98, which used BP. Distilled water served as the negative control. Three replicate plates were prepared for each test condition. After exposure for 20 min at 37 °C, and incubation for 48 h at 37 °C, the number of colonies per plate was counted manually with an electronic register (Model 570, SUNTEX, Taipei, Taiwan). The test result was recorded as experimental value, average value, and standard deviation for the number of revertant colonies per plate. The result was deemed “positive” if there was a dose-dependent increase and/or a reproducible increase at one or more concentrations in the number of revertant colonies per plate in at least one strain with or without metabolic activation.
Chromosomal aberration assay
Chinese Hamster Ovary cells (CHO-k1) were obtained from the Korean Cell Line Bank (KCLB, Seoul, Korea). The cells were cultured in F12 nutrient mixture (GIBCO, Grand Island, NY, USA) and 10% fetal bovine serum (Corning, NY, USA) at 37 °C and 5% CO2 and were subcultured every 3–4 days. The S9 preparation used for metabolic activation is described under the methods for the bacterial reverse mutation assay.
A preliminary dose range finding study was performed with eight concentrations up to 5000 µg/mL to determine the highest concentration used for each exposure scenario (24 h incubation without S9 and 6 h treatment and 18 h recovery with or without S9). For the 24-h incubation scenario, the relative increase in cell counts (RICC) was 46.40% and 60.39% in for 185.19 µg/mL and 61.73 µg/mL, respectively. For the 6 h exposure group without S9 mix, RICC were 46.77% and 61.55% for 555.56 µg/mL and 185.19 µg/mL, respectively. For the 6 h exposure group with S9 mix, RICC were 49.64% and 61.37% for 1666.67 µg/mL and 555.56 µg/mL, respectively. Based on these results, the maximum concentrations chosen for use in the chromosome aberration study were 185.19 µg/mL for 24 h, 555.56 µg/mL for 6 h-S9 and 1666.67 µg/mL for 6 h + S9. Two lower concentrations were also used in each assay, at threefold dilutions from the highest concentrations.
For the assay, the positive control substance was mitomycin C without metabolic activation (S9 mix), and cyclophosphamide with S9 mix. The negative control was distilled water. Approximately 22 h after treatment, colcemid (GIBCO) was added to each culture plate for a final concentration of 0.2 µg/mL. The cultures were incubated for an additional 2 h, after which they were detached using 1X trypsin solution. The medium containing mitotic cells was centrifuged at 1000 rpm for 5 min. The cell pellets were resuspended in 75 mM potassium chloride solution and incubated at 37 °C for 20 min. The cells were fixed 3 times with Carnoy’s fixative solution (acetic acid:methanol = 1:3 v/v) for slide preparation. The slides were stained with 5% Giemsa solution for 5 min and observed microscopically. Two slide samples were prepared from each plate and 150 metaphase cells per plate were counted.
Structural (gaps, breakage, exchange) and numerical aberrations were evaluated. Aneuploidy was not counted as numerical aberration since it occurs frequently in cell-lines such as CHO-k1. Endoreduplication was classified as polyploidy, and it was recorded if endoreduplication was frequently observed. The test material was considered to be clearly positive for clastogenicity (and/or aneugenicity) if at least one of the test concentrations exhibited a statistically significant increase compared with the corresponding negative control, the increase was dose-related and any of the results were outside of the distribution of historical negative control data.
Micronucleus assay
The study was conducted to evaluate the potential genotoxicity of the test material in an in vivo micronucleus assay. Male SPF CrlOri:CD1 (ICR) mice (n = 25) from Orient Bio Co., Ltd were used and divided into five equal groups: one negative control, one positive control and three treatment groups (1250, 2500 and 5000 mg/kg bw/day). The positive control used in the study was mitomycin C (2.0 mg/kg bw/day) and the negative/vehicle control was distilled water. Animals were dosed with the test material orally once a day for two days at 24-h intervals after being subjected to a 3–4 h fast, Mitomycin C was administered intraperitoneally rather than orally. Animals were observed on the day of administration from 30 min to 4 h after administration and at the time of autopsy for the occurrence of dead animals and abnormal signs. All animals were weighed before acquisition and grouping, administration of test substances, and before bone marrow collection. At 18 and 24 h after the last dose, all animals were euthanized, and bone marrow was collected from the thigh bone and suspended in fetal bovine serum. Bone marrow smears (3/animal) were prepared on glass slides at room temperature and fixed in methanol for 5 min. To score the polychromatic erythrocytes (PCE) to normochromatic erythrocytes (NCE) ratio, slides were stained with 4% Giemsa solution. To score the micronucleated polychromatic erythrocytes (MNPCE) from PCEs, the slides were fixed, stained with acridine orange, and covered with cover glasses.
The observation of slides was performed with the investigator blinded to test condition. The PCE to NCE ratio was determined by optical microscope at greater than 1000 × magnification. MNPCEs were observed by fluorescence microscope equipped with an FITC filter and more than 400 × magnification. The PCE/NCE ratio was determined by scoring the number of PCEs and NCEs in 500 erythrocytes per animal. Micronucleus frequency was determined by analyzing the number MNPCEs from 4000 PCEs per animal. Under the fluorescence microscope, PCEs appeared as a red-fluorescent and did not stain for nuclei using acridine orange. NCEs appeared only as shadow entities without fluorescence. With the optical microscope, PCEs stained with Giemsa appeared blue or purple, and NCEs appeared pink. In order to judge the presence of micronuclei, the largest size was defined as ½ the size of erythrocyte diameter and the smallest size was the limit of identification. Shapes included circle, donut, and semi-circle and color was defined as the same as the nucleus of the cell.
The result was considered positive if all of the following acceptability criteria were met: (1) the frequency of MNPCE/4000 PCEs (mean ± SD, %) is statistically reliable, increases dose-dependently, and/or shows a reproducible positive reaction at one or more concentrations, and (2) any of the results are outside the distribution of historical negative control data.
Statistical analysis
The statistical package SPSS 12.0 K (SPSS, Chicago, IL, USA) was used for all analyses. The criterion for significance was p < 0.05. In the single dose oral toxicity study, body weight data were analyzed using the one-way analysis of variance (ANOVA) test to detect any differences between groups. In the 14-day dose range finding study, differences between groups were examined by the one-way ANOVA test. If a significant difference was detected, the Duncan test was used for data comparison if there was homogeneity of variance, and Dunnett’s T test was used if there was not homogeneity of variance. In the 13-week toxicity study, continuous data were compared using the one-way ANOVA test to determine if there was a difference between treated animals and controls. If there was a significant difference, the data were analyzed similarly to the 14-day study. Detailed clinical signs, urinalysis, urine sediments, and estrus cycle data were analyzed as non-continuous data and were converted by scale conversion and analyzed by the Chi-squared test. In the chromosome aberration assay, the number of aberrant metaphases, excluding gaps, and the incidence of polyploid plus endoreduplication were analyzed. Comparisons between groups were performed using the Chi-squared test. A linear logistic regression test was used to determine if the response was dose dependent. In the micronucleus assay, the negative control and treatment groups were compared by one-way ANOVA. If a significant difference was detected, the data were analyzed similarly to the 14-day study.








留言 (0)