
記住我



Duchenne muscular dystrophy (DMD) is the most common muscular dystrophy affecting children. With an incidence between 1:3500-1:5000 live male births1, the high frequency of de-novo mutations in the X-linked DMD gene is an obstacle to its eradication.
The DMD neuromuscular and cardiac manifestations are well recognised: affected children are diagnosed in the first few years of life and weakness progresses rapidly, leading to loss of ability to walk by the early teens, and subsequent respiratory and cardiac insufficiency resulting in shortened life span. The course is secondary to the loss of dystrophin, a crucial protein that protects the sarcolemma from contraction-induced damage leading to progressive muscle fibres loss. In the last decade, a few pharmacological and genetic therapies that reduce muscle damage have demonstrated efficacy in clinical trials and are at different stages of approval2.
However, DMD is much more than a pure neuromuscular disease: already in 1868, the French neurologist Duchenne De Boulogne in his original report of 13 boys with DMD indicated that six of them had a low IQ, two had language problems and another two had epilepsy3.
These observations have been confirmed in subsequent studies reporting that nearly half of individuals affected by DMD may have a complex neurobehavioural and neurocognitive phenotype. This can variably manifest with specific language disorders, intellectual disability, reading disorder, autism spectrum disorder (ASD), attention-deficit/hyperactivity disorder (ADHD), emotional disorders and obsessive-compulsive disorders (OCD)4,5,6.
Work performed in the last decade has shed further light on the variable brain involvement: this largely relates to the location of the DMD mutation along this large gene. We are now aware that the DMD locus produces multiple dystrophin isoforms, which can be differentially affected depending on where the pathogenic variant is located7.
More recent work has improved the understanding of the role of individual isoforms on the brain phenotypes observed, both in the human and mouse models; and on the biological role of these dystrophins8.
Ongoing work is exploring the role of genetic therapy administration, not to the muscle but to the brain, in improving the brain comorbidities in animal models of DMD, potentially paving the way to their future root cause correction.
While these basic research efforts continue, improvement in the screening, assessment, and diagnosis of the brain comorbidities in patients with DMD need to be implemented, together with a systematic assessment of the role that psychotherapy and psychopharmaca may play in managing some of the neurobehavioural consequences. This is not sufficiently addressed by the current standards of care for DMD9, and continues to be a major and growing unmet need for this patient population, especially as life expectancy increases.
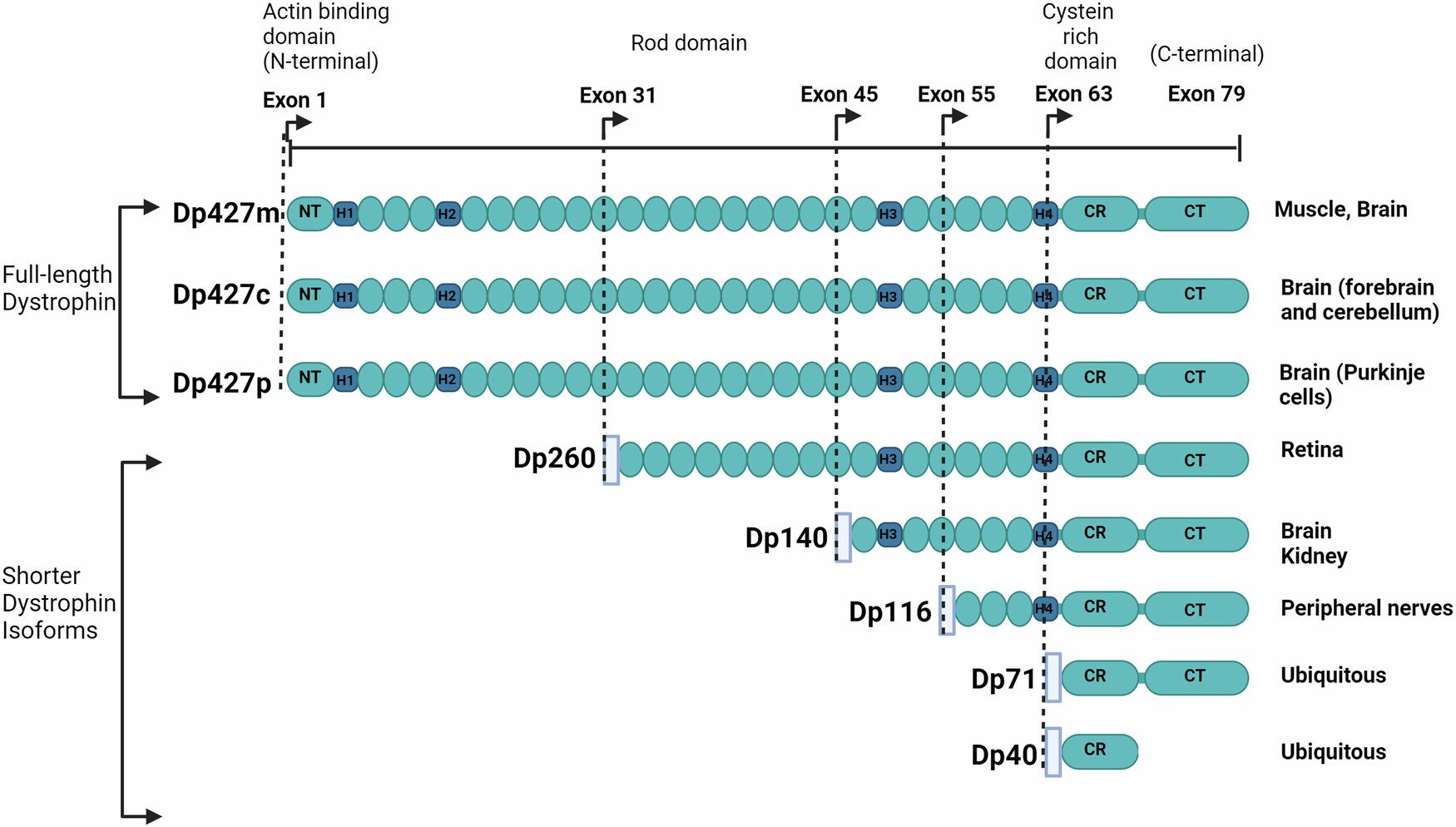
The DMD locus, brain DMD isoforms and their pattern of expressionThe Xp21.1-p21.2 DMD gene, the largest gene in the human genome, spans nearly 2.5 Mb. It consists of 79 exons and seven internal promoters, each linked to unique first exons giving rise to at least 7 protein products of different sizes7. The full-length isoform (Dp427) is transcribed through 3 different tissue-specific promoters upstream of their first exon sequences, namely Purkinje (P), muscle (M), and cortical (C); they are expressed in different cell types and/or cell compartments in association with different molecular partners, as indicated by transcriptomic studies from Allen Human Brain and BrainSpan atlases (https://atlas.brain-map.org; http://www.brainspan.org) and mouse tissue8,10. While Dp427m is mainly expressed in muscles, likely including smooth muscles of brain arterioles, Dp427c is mainly present in the forebrain and cerebellar neurons. The shorter isoforms named according to their molecular weight, Dp260, Dp140, Dp116, Dp71, and Dp40, are produced by independent downstream promoters and exhibit tissue-specific expression patterns as shown in studies on murine and canine embryos and PC12 cell lines11,12 (Fig. 1). Of note, some of these isoforms also express different alternative splicing that may regulate their subcellular location12. While Dp260 and Dp116 are expressed in the retina and peripheral nerve, respectively, transcriptomic analysis from Allen Human Brain and BrainSpan atlases demonstrated that Dp140 and Dp71/Dp40 are expressed in the brain with larger expression in hippocampus and amygdala8. Dp71 and Dp140 are also expressed outside the central nervous system (CNS). In particular, Dp71 is expressed in many human adult tissues including liver, lung, kidney, and muscle progenitor cells, and skeletal muscle13,14, while Dp140 is also transiently expressed during kidney development15.
Fig. 1: DMD gene organization and associated protein products.
The full-length dystrophins (Dp427m,c,p) have a modular structure containing the N-terminus (NT) with F-actin binding sites, an extensive central rod domain that consists of β-spectrin-like repeats (oval shape symbols) with binding sites for F-actin, sarcolemma and multiple cellular proteins, and proline-rich hinge regions (H1-H4) predicted to form triple-helical coiled coils, followed by cysteine-rich (CR) and C-terminal (CT) domains. The shorter dystrophin isoforms (Dp260, Dp140, Dp116, Dp71) exhibit at least the cysteine-rich (CR) and C-terminal (CT) domains, except Dp40 that share the Dp71 first exon but misses the CT domain due to an alternative polyadenylation that generates a stop codon. The CR region contains a WW domain, two EF-hands, a β−dystroglycan binding site, and one ZZ domain. The C-terminal region contains two syntrophin binding sites and a coiled-coil domain interacting with dystrobrevins. The light blue rectangular boxes indicate the position of the promoter region of each isoform. The main tissues expressing the distinct dystrophin isoforms are indicated on the right. The figure was Created in BioRender101.
The expression of these dystrophin isoforms changes during development. Dp140 is more abundant in the foetal brain; Dp427c and to a less extent Dp427m are more highly expressed in adults compared to foetal brain, while Dp71/Dp40 expression is high both during foetal stages and later in human life8. More recently it has been found that Dp427p was absent throughout the development of the brain as shown in transcriptomic analysis from Allen Human Brain and BrainSpan atlases, which is contrast with previous studies8,16.
Pathogenic DMD gene variants, more frequently out-of-frame deletions (65%) or duplication (15%), lead to the absence of dystrophin in muscle resulting in DMD. Allelic variants, often in-frame deletions, can also induce the production of a partially functional dystrophin protein and cause the milder and later onset Becker muscular dystrophy (BMD). The location of the mutation along the DMD gene may differentially impact the multiple dystrophin isoforms (Fig. 1). While all mutations affect muscle and brain production of Dp427, the additional involvement of Dp140 and/or Dp71/40 is related to how further downstream in the DMD gene the mutations are located (Fig. 1).
Role of the different isoforms in the brainRecent advances in cellular and molecular neuroscience have shed light on the pivotal role of dystrophin isoforms in modulating synaptic transmission and ion channel functioning. The Dp427 isoforms have been implicated in the localisation and functioning of central γ-Aminobutyric acid type A (GABAA) receptors, which has been indirectly confirmed by brain imaging in adults with DMD, although it is not clear if all three full length isoforms contribute to the GABA synaptic functioning17. Immunofluorescence studies in the dystrophic mdx mice carrying a nonsense mutation in exon 23 have demonstrated that the Dp427 deficiency reduces the expression of GABAA receptors in key brain regions, including the amygdala, hippocampus, cerebral cortex, and cerebellum18,19. This disrupts GABAergic inhibitory postsynaptic currents, particularly in the neurons of the basolateral nucleus of the amygdala in response to norepinephrine inputs, which has been associated with abnormal defensive/fear and anxiety behaviours. In hippocampus, altered GABAergic inhibition is posited to enhance NMDA receptor-dependent synaptic plasticity and impacts memory consolidation, as well as synaptic plasticity in cerebellum, with putative impact on both motor and cognitive functions19,20,21,22,23. Recent behavioural studies in mdx mouse models highlighted that Dp427 deficiency may alter the subunit composition of GABAA receptor subtypes at both synaptic and extrasynaptic sites, with a differential impact on distinct brain structures24,25. Using a different strain of dystrophic mice (DBA/2 J mdx mouse), and a different stimulation protocol, Bianchi et al could not fully replicate the findings on synaptic plasticity, highlighting the importance of standardising the experimental procedures related to these assessments26. As these mouse models are deficient in all three Dp427 isoforms, it is not possible to conclude from these studies which one is relevant for the observed phenotypes, or if they all contribute to this.
Regarding Dp140, gene ontology analysis suggested a role during the early stages of neurodevelopment8. The combined absence of both Dp427 and Dp140 in the exon52-deleted mdx52 mouse model has been associated with abnormal social behaviours and diminished glutamatergic transmission, paralleling the ASD-like symptoms of DMD children lacking this isoform27,28. A recent study demonstrated a direct interaction of Dp140 with neuronal voltage-gated Ca2+ channels of the CaV2 subfamily in the mouse brain, using immunoprecipitation and proximity ligation assay techniques, thus providing new avenues for understanding the neurobiology of cognitive dysfunctions in DMD29.
Dp71, the most abundant isoform in the brain, plays a crucial role in the functioning of astrocytes, as demonstrated in mouse brain and human astrocytes’ studies using immunofluorescence and immunocytochemistry techniques30,31. Figure 2 provides a schematic representation of the different brain protein complexes involved with the different dystrophin isoforms.
Fig. 2: Schematic Representations of Dystrophin Interactions in the Nervous System.
A Dp427 and Postsynaptic Complexes in Central Inhibitory Synapses. This diagram illustrates the Dp427-associated complex, including α and β dystroglycans, and its role in postsynaptic scaffolding (the black U-shape form represents dystrobrevin). The interaction with key proteins such as Neuroligin-2 (NLGN2), neurexin, S-SCAM, and IQsec3, which are crucial for GABAAR clustering, is depicted. The GABAAR subunits are colour-coded to identify α subunits in light blue, β subunits in purple, and γ2 subunits in red24. B Dp140 complexes in Synapses. This illustration shows the Dp140 complex associated with Cav2.1 and glutamate axis. C Dp71 and its Association with AQP4 and Kir4.1 Channels in Astrocyte Endfeet. This panel demonstrates the interaction of Dp71 with AQP4 and Kir4.1 channels within the perivascular astrocyte endfeet, indicating its potential role in neurovascular coupling102. α-DG α-dystroglycan, β-DG β-dystroglycan, nNOS neuronal nitric oxide synthase, NLGN2 neuroligin 2, IQsec3 gephyrin-associated IQ motif and SEC7 domain-containing protein 3 (also called SynArfGEF), S-SCAM synaptic scaffolding molecule (also called MAGI-2 for membrane-associated guanylate kinase inverted 2); CaV2.1 calcium channels Kir: potassium channels, AQP-4 aquaporin-4, α-Dbv-1 α-dystrobrevin-1, DG dystroglycan, Syn syntrophin. Created in BioRender103.
Genotype/phenotype correlations for DMD patients with mutations affecting the different DMD isoformsSeveral studies reported the prevalence of brain related comorbidities in DMD, suggesting a role for the site of mutations and their effect on the expression of the different brain dystrophin isoforms. There is a large variability in the reported prevalence of the individual brain related symptoms due to differences in sample sizes, assessment instruments, and data collection methods4,32,33.
Assessment of neurocognitive and neurobehavioral functioning can be complicated in patients with DMD because of severe motor impairments and consequentially limited social interactions. Normative instruments being used are based on non-motor impaired persons and DMD-specific instruments are not available. However, patients with other severe neuromuscular conditions, such as Spinal Muscular Atrophy34, glycogen storage disorders, neuropathies and muscular dystrophies due to involvement of dystrophin complex members exclusively expressed in muscle35 are not associated with the brain comorbidities observed in DMD. Furthermore, the behavioural and neurocognitive defects in the form of autistic spectrum disorder can often manifest before the occurrence of clear muscle weakness in DMD36.
This, together with clear genotype/phenotype/brain isoforms correlation and the supportive evidence from dystrophic animal models, strongly supports the view that CNS involvement in DMD is a primary consequence of brain dystrophin deficiency, not a secondary phenomenon. Furthermore, in clinical practice and scientific research normative instruments such as the Wechsler intelligence scales prove to be useful33,37. An important challenge for future research is to make a DMD specific and sensitive battery of instruments based on currently available instruments.
Despite these methodological considerations, there is overall consistency in phenotype/genotype correlation concerning specific pathological aspects:
Early development. Developmental milestones in DMD children demonstrate that the milestones requiring more cortical involvement, such as sitting or walking, are typically delayed38, with a gradient that directly correlates with the number of isoforms affected in the CNS and the associated cognitive function39,40. Studies using the Griffiths and the Baileys neurodevelopmental scales suggest that DMD children have a developmental quotient that is on average 1 standard deviation below population mean41. The scores in the subscales such as language, personal, social and eye and hand coordination, are lower in patients with involvement of Dp140 and Dp7141.
Motor function in relation to CNS involvement. Several studies using the North Star Ambulatory Assessment (NSAA) motor function scale that also includes timed items, have demonstrated significantly lower peak achievement in children lacking Dp140 and even lower in those also lacking Dp7139,40,42,43,44. Importantly similar differences in motor function can also be observed in the different mdx mouse models lacking different dystrophin isoforms39.
Intellectual functioning, cognitive and academic abilities. A recent meta-analysis of 32 studies using the Wechsler intelligence scale (preschool, child and adult studies) in 1234 DMD individuals found that the mean full-scale IQ is approximately 1 standard deviation below average norm, i.e full-scale IQ of 84.7633. A review of the global prevalence of intellectual developmental disorder, operationalized as a full-scale IQ of less than 70 (2 standard deviations below the mean of 100), was found in 22% of DMD patients with lower prevalence (12%) in patients with exclusive involvement of Dp427, and 2.5 and 6 fold higher prevalence when Dp140 and Dp71 were also involved45. Neurocognitive functions such as working memory and executive functions (inhibition, switching, problem solving and planning) are often impaired even in boys with a normal full-scale IQ46,47. Other studies have specifically assessed academic abilities, demonstrating a higher prevalence of reading disabilities46,48,49,50 and reduced arithmetic skills51. Academic performances were also lower for mutations affecting both Dp140 and Dp7146. Longitudinal studies have shown that using the Wechsler scales the full-scale IQ is consistent over time37. However, DMD boys with attention and behavioural disorders often had less consistent results with larger full-scale IQ changes over consecutive assessments.
Neuropsychiatric comorbidities. Prevalence rates of these disorders are significantly higher than in the general population. There is, however, a large degree of heterogeneity in reported prevalence rates over different studies32 with prevalence of 0.00 indicating a 0% prevalence and prevalence of 1.00 indicating a 100% prevalence: ASD (13 studies; 0.01 to 0.21 with a mean of 0.06 and 0,0076 in general population), ADHD (10 studies; 0.03 to 0.50 with a mean of 0.18 and 0,034 in general population) and OCD (6 studies; 0.05 to 0.33 with a mean 0.12 and 0,012 in general population). Interestingly, data on the milder BMD variant describes comparable prevalence rates: ASD 0.06, ADHD 0.28 and OCD 0.0732.
While intellectual function is clearly linked to the integrity of the two shorter Dp140 and Dp71 isoforms, the correlation for the behavioural comorbidities is less consistently linked to these isoforms. Indeed, neuropsychiatric aspects such as ASD and to a certain extent also ADHD, can be found across the spectrum of mutations, pointing towards a role for Dp4274,32,52,53. A recent metanalysis has nevertheless suggested that ADHD might be more often associated with boys lacking all the isoforms, i.e. Dp427, Dp140 and Dp7132.
Other studies have more specifically addressed anxiety and fear-based disorders (such as social and separation anxiety, panic disorder and specific phobias) that are reported in 24% to 33% of people with DMD54, hence more common than in the general population5,55,56. Obtaining objective measures of anxiety using behavioural startle responses, children with DMD presented increased unconditioned startle responses to threats. This pathological response is a feature of all DMD children hence likely related to lack of the Dp427 isoforms; in addition, there was evidence that the additional involvement of the Dp140 isoform further increased the abnormal startle response and the risk of anxiety56. Figure 3 gives a schematic representation of ten areas of brain related comorbidities (big ten of Duchenne). It is important to recognise that in clinical practice there may be a complex overlap of these areas. This complexity may hinder adequate screening and assessment57.
Fig. 3: Domains and areas of brain function affected in DMD.
Systematic overview of brain related comorbidities in DMD (Big ten of Duchenne) representing four domains and ten areas of (dys)functioning: A neurocognitive (executive functions, working memory and intellectual functioning); B Academics (reading and arithmetic); C emotional: (anxiety and depression; D neuropsychiatric (autism, Attention-Deficit Hyperactivity Disorder: ADHD, Obsessive Compulsive Disorder: OCD).
Brain Imaging. Brain magnetic resonance imaging (MRI) reveals no structural abnormalities; however total brain and grey matter volumes are reduced compared to healthy controls58. Measurements of the white matter show lower fractional anisotropy, and higher diffusivity in DMD children than in healthy controls. Genotype/phenotype correlation revealed that children lacking both Dp427 and Dp140 had more obvious grey matter volume differences and reduced cerebral blood flow compared to those with involvement of Dp427 only58. The connectomic disturbances have been confirmed by tractography and functional MRI studies using a resting state and suggest an over-activation of the default mode network and executive control network, with suppression of primary sensorimotor cortex and cerebellum-visual circuit58,59. The combination of functional and structural MRI together with neuropsychological tests has also allowed one to collect information on the relationship between function and structures and the involvement of neural networks including the cerebellar-thalamo-cortical loop. The pathological white matter connectivity and fibre organization in cerebellar tracts may both contribute to neurocognitive dysfunctions observed in children with DMD59,60,61,
留言 (0)