
記住我
Traditionally, bioinformatics analyses often utilize the differential gene abundance between tumor and normal tissues to screen for target genes (Golub et al., 1999). Typically, genes with significantly higher abundance in tumors than normal tissues are classified as oncogenes and become focal points of research (Bishop, 1991; Croce, 2008). However, a new understanding has emerged with the proliferation of comprehensive databases such as TCGA. Researchers increasingly recognize that not all genes are highly expressed in tumors act as promoters of cancer (Cao et al., 2021; Yan et al., 2019; Boor et al., 2020; Martinez-Turtos et al., 2022). In fact, some genes show high abundance in tumor tissues but are associated with good patients prognosis (Cao et al., 2021; Yan et al., 2019; Boor et al., 2020; Martinez-Turtos et al., 2022). This finding challenges the traditional understanding of tumor biology. Although public databases provide evidence that genes which are highly expressed in tumor tissues and serve tumor suppressor roles are prevalent, they have not been broadly recognized or systematically analyzed by the scientific community. Our work pioneers the classification and definition of these genes as “paradoxical genes,” and it delves deeply into the reasons and context for the existence of these paradoxical genes.
The discrepancy between mRNA and protein levels may be a reason for the emergence of paradoxical genes (Vogel and Marcotte, 2012). This difference is mainly due to post-transcriptional and -translational modifications, which are crucial in the dynamic regulation of gene expression (Vogel and Marcotte, 2012). Post-transcriptional modifications include processes such as alternative splicing, enabling a single gene to generate multiple mRNA variants, thereby expanding the diversity of the proteome (Vogel and Marcotte, 2012; Wahl et al., 2009). The upstream open reading frames (uORFs) can significantly regulate the translation of the main open reading frame (ORF) (Barbosa et al., 2013; Johnstone et al., 2016). Concurrently, post-translational modifications such as phosphorylation and ubiquitination further diversify protein functions and regulation (Hershko and Ciechanover, 1998; Hunter, 2007). However, within the context of TCGA, the focus is solely on the measurement of mRNA abundance (Cancer Genome Atlas Research Network, 2008; Author anonymous, 2012; Weinstein et al., 2013). This is typically quantified using RNA sequencing data, which provides detailed information on the levels of mRNA present in a given sample (Cancer Genome Atlas Research Network, 2008; Author anonymous, 2012; Weinstein et al., 2013). Inconsistencies between gene and protein levels can potentially distort patient prognosis results, contributing to the emergence of paradoxical genes (Akbani et al., 2014).
The tumor immune microenvironment (TIME) is critical for suppressing tumor progression, through its complex network of immune cells, stromal cells, signaling molecules, and extracellular matrix components (Joyce and Fearon, 2015; Fridman et al., 2012; Quail and Joyce, 2013). This dynamic environment can promote or inhibit tumor growth, depending on the balance of pro- and anti-tumor factors (Schreiber et al., 2011). Key players include cytotoxic T lymphocytes (CTLs), natural killer (NK) cells, dendritic cells, B cells, proinflammatory cytokines, and chemokines (Schreiber et al., 2011; Smyth et al., 2002; Palucka and Banchereau, 2012; Nelson, 2010; Müller et al., 2009; Topalian et al., 2015). There is currently evidence that some Paradoxical genes are involved in regulating TIME and inhibiting tumor progression (Cao et al., 2021; Yan et al., 2019; Boor et al., 2020; Martinez-Turtos et al., 2022). The regulatory influence of paradoxical genes on TIME elucidates the mechanism underlying their tumor suppressor effects.
The expression of genes and pathways exhibits context-dependent effects, also known as context specificity, which refer to the phenomenon where the function or behavior of a gene varies depending on the specific context in which it operates (Feil and Fraga, 2012; Huang, 2009; Beyer et al., 2007). In the context of cancer, the role of a gene can vary significantly depending on factors such as the type of cancer, the tissue in which the tumor originates, and the stage of the cancer (Vogelstein and Kinzler, 2004; Hanahan and Weinberg, 2011; Stratton et al., 2009). For instance, genes associated with signaling pathways like TGFβ, NOTCH, and NF-κB demonstrate differential expression across various tumor tissues. The TGFβ pathway presents a dual role, acting as a tumor suppressor in early stages and a promoter in advanced stages, with the expression varying significantly in breast, pancreatic, and colorectal cancers (Massagué, 2008). Similarly, the NOTCH signaling pathway, critical for cell fate determination and differentiation, shows oncogenic as well as tumor-suppressive functions, depending on the cancer type, as observed in breast cancer (BRCA), T-cell acute lymphoblastic leukemia, and lung cancer (Stylianou et al., 2006; Weng et al., 2004; Kopan and Ilagan, 2009). Often, the NF-κB pathway, a key regulator of inflammation and immune responses, is dysregulated in BRCA, multiple myeloma, and colorectal cancer, where it facilitates cell proliferation, survival, and chronic inflammation (Karin, 2006; Annunziata et al., 2007; Greten et al., 2004). The specific roles and expression patterns of these similar pathways in different tumor environments are also one of the reasons for the widespread existence of paradoxical genes.
In this article, by exploring the mechanism of paradoxical genes formation, we seek to broaden the current understanding of tumor biology and provide new ideas for tumor treatment and research.
2 Paradoxical genes emerge as a key focus in bioinformatics researchWhen genes highly expressed in cancer compared to normal tissues, are identified through bioinformatics analyses, it often implies that these genes may act as drivers of oncogenesis, serve as diagnostic or prognostic biomarkers, or act as predictive markers for treatment response (Niu et al., 2022; Liu Y. et al., 2021). Indeed, the relationship between gene expression and cancer prognosis can be complex and sometimes counterintuitive (Hanahan and Weinberg, 2011). Contrary to what might be expected, high expression of certain genes in tumors can be associated with a more favorable prognosis (Hanahan and Weinberg, 2011). This paradoxical finding highlights the complexity of cancer biology, revealing that genes may play multifaceted roles in tumorigenesis and cancer progression (Hanahan and Weinberg, 2011). For instance, some genes highly expressed in tumors could participate in immune response activation, DNA repair mechanisms, or cellular differentiation processes, potentially inhibiting tumor growth or spread, thereby improving patient outcomes (Blagih et al., 2020; Williams and Schumacher, 2016; Kalluri and Weinberg, 2009). Researchers face a challenge of dissecting the dual roles that some genes, acting as oncogenes in certain contexts while serving protective or suppressive functions in others (Hanahan and Weinberg, 2011).
2.1 Paradoxical genes exhibit differential and uneven expression across various tumorsIn recent years, our understanding of the molecular underpinnings of cancer has been revolutionized by the integration of genomic databases such as TCGA into cancer research (Tomczak et al., 2015). TCGA provides an extensive compilation of genetic mutations, gene expression data, and epigenetic alterations across thousands of tumors, spanning over 30 human tumor types (Weinstein et al., 2013; Tomczak et al., 2015). Through our analysis of the TCGA database, our team has determined that the role of paradoxical genes cannot be overlooked. We conducted a detailed analysis of the distribution of paradoxical genes across various tumor types, ranking them by the number of highly expressed genes within each tumor category (Figure 1A). We observed that this type of gene is ubiquitously present across various cancers. Notably, BRCA, bladder urothelial carcinoma, and kidney renal clear cell carcinoma (KIRC) exhibit the highest expression levels of these tumor suppressor genes. In contrast, kidney chromophobe (KICH), esophageal carcinoma (ESCA), and prostate adenocarcinoma (PRAD) have significantly lower expression of these genes.
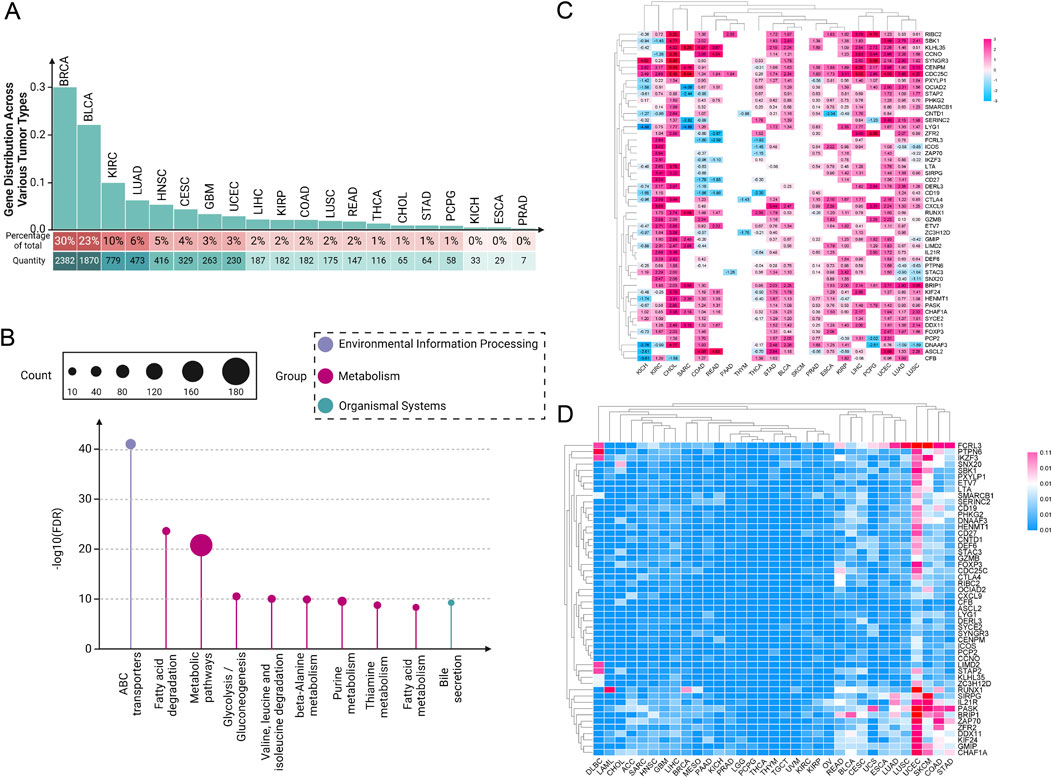
Figure 1. Pan-cancer analysis of paradoxical genes. (A) Expression of paradoxical genes in various tumors. The proportion of paradoxical genes in tumors is rounded to the nearest whole number; (B) KEGG analysis of 254 paradoxical genes; (C) Heat map depicting expression intensity of 50 paradoxical genes across various tumors; (D) SNV of paradoxical genes.
This disparity suggests a complex regulatory mechanism involved in the expression of paradoxical genes, which the tumor microenvironment (TME) and specific oncogenic pathways could influence. The high expression levels of paradoxical genes in BRCA, BLCA, and KIRC suggest that these cancers possibly utilize these genes to balance between tumor suppression and oncogenic activity, potentially as a response to oncogenic stress or other cellular pressures. In contrast, the reduced expression of paradoxical genes in KICH, ESCA, and PRAD might indicate a loss of this balancing mechanism, possibly contributing to more aggressive tumor behavior. These findings provide a crucial direction for future research into the mechanisms regulating paradoxical genes and their role in cancer progression.
2.2 Kyoto encyclopedia of genes and genomes analysis of paradoxical genes: insights into their relationship with tumor metabolismTo investigate the functional mechanisms of paradoxical genes further, we initially screened 254 paradoxical genes for a KEGG analysis (Figure 1B). A common characteristic among these genes is their high expression in at least three tumor cell groups, correlating with improved patient prognosis. Our KEGG analysis revealed that these genes are extensively involved in various metabolism-related pathways, suggesting it as a primary mechanism through which paradoxical genes influence tumor prognosis.
Notably, liver X receptor (LXR) genes, including LXRα and LXRβ, are a few examples of this phenomenon (Han et al., 2023; Wang et al., 2023). LXRs are nuclear receptors involved in lipid metabolism, inflammation, and cholesterol homeostasis (Zelcer and Tontonoz, 2006). In the context of cancer, LXRs have demonstrated a dual role in tumor prognosis, influenced by their regulatory impact on metabolic pathways and immune responses in the tumor microenvironment (Han et al., 2023; Wang et al., 2023). LXR genes can play a role in inhibiting tumor progression (Zhang et al., 2014; Nguyen-Vu et al., 2013). This is primarily mediated through their anti-inflammatory effects within the tumor microenvironment. High expression of LXR genes causes upregulation of cholesterol efflux transporters such as ATP-binding cassette transporter A1 (ABCA1) and ABCG1, which facilitate cholesterol efflux and reduce lipid accumulation within macrophages, thus attenuating the inflammatory response associated with tumor progression (Joseph et al., 2003; Wang et al., 2006). Furthermore, LXR activation has been linked to the suppression of inflammatory cytokine production by immune cells, leading to a less conducive environment for tumor growth (Joseph et al., 2003; Fessler, 2016; Chawla et al., 2001). A study demonstrated that LXRs activation disrupts BRCA cell proliferation by downregulating the expression of genes involved in cell growth and proliferation, particularly those regulated by the E2F family of transcription factors (Nguyen-Vu et al., 2013). The activation of LXRs leads to the downregulation of key genes involved in the cell cycle, DNA replication, and other critical processes for cancer cell division (Nguyen-Vu et al., 2013). This effect is partly mediated through the regulation of E2F2, highlighting a potential mechanism by which LXRs inhibit proliferation in cancer cells (Nguyen-Vu et al., 2013).
Conversely, LXRs can promote tumor growth through several mechanisms. Their activation leads to upregulation of genes involved in lipid biosynthesis, such as SREBP-1c (Sterol Regulatory Element-Binding Protein 1c) (Okazaki et al., 2010; Jeong et al., 2021). SREBP-1c is a crucial transcription factor that enhances the expression of genes required for fatty acid and triglyceride synthesis (Okazaki et al., 2010; Jeong et al., 2021). In many cancers, particularly those with high lipid requirements like breast cancer, this can contribute to tumor cell proliferation and survival by ensuring a steady supply of essential lipids that are critical for membrane synthesis, energy storage, and signaling (Santos and Schulze, 2012; Menendez and Lupu, 2007; Swinnen et al., 2006; Zadra et al., 2013; Fukuchi et al., 2004; Vedin et al., 2009). Our research (Figure 1A) demonstrated that paradoxical genes are prevalently expressed in breast cancer, which also supports the notion that the modulation of these genes, particularly their regulation of tumor lipid metabolism, is fundamental to their anticancer effects.
In our previous investigation of clear cell renal cell carcinoma (ccRCC), we further examined the dual role of LXR (Wu et al., 2019). Our findings suggest that LXR functions as a balance gene, where both heightened and diminished expression levels can exert inhibitory effects on ccRCC progression (Wu et al., 2019). Specifically, both LXR agonists and inverse agonists inhibits cell proliferation and colony formation. The LXR agonist LXR623 downregulates low-density lipoprotein receptor (LDLR) and upregulated ABCA1, causing a decline in intracellular cholesterol and induced apoptosis (Wu et al., 2019). Conversely, the LXR inverse agonist SR9243 downregulates key FA synthesis proteins, including sterol regulatory SREBP-1c, FA synthase, and stearoyl-CoA desaturase 1(SCD1), leading to decreased FA content and apoptosis in ccRCC(Wu et al., 2019). This phenomenon illustrates that alterations in cancer metabolism are a pivotal factor in mediating the regulatory effects of paradoxical genes on tumor prognosis.
2.3 Expression intensity and single nucleotide variations (SNV) analysis of paradoxical genes across different tumorsSubsequently, we screened out 50 paradoxical genes for pan-cancer analysis (Figure 1C). Their common feature is that they are highly expressed in ≥4 groups of tumor cells, and are associated with better patient prognosis. The expression intensity of these genes was analyzed in 20 different tumor tissues, and we found widespread overexpression, including, but not limited to, KIRC, cholangiocarcinoma (CHOL), stomach adenocarcinoma (STAD), BLCA, PRAD, ESCA, and liver hepatocellular carcinoma (LIHC), highlighting their potential as pan-cancer prognostic markers. Interestingly, subsequent analysis of SNVs in these genes showed that mutations are infrequent, a characteristic not generally observed in traditional oncogenes (Figure 1D). This observation further supports the hypothesis that the prognostic effect of paradoxical genes is mediated through mechanisms distinct from those employed by oncogenes, potentially driving tumor evolution towards less aggressive and more treatable forms. Nonetheless, there are currently very few reports on the occurrence of highly expressed tumor suppressor genes in tumors. Uncovering previously underappreciated complexities in the relationship between gene expression and cancer prognosis is critical.
3 The relationship between gene abundance and protein abundance: is there always a direct correlation?The central dogma of molecular biology, formulated by Francis Crick, describes the flow of genetic information from DNA to RNA to protein through the processes of transcription and translation (Crick, 1970). While it is generally hypothesized that higher mRNA levels correlate with higher protein levels, this relationship is influenced by several factors (Vogel and Marcotte, 2012; Schwanhäusser et al., 2011; Tian et al., 2004). Post-transcriptional regulation can modify mRNA stability and translation efficiency, as well as the sequence features of the mRNA itself, such as upstream ORFs, can affect how efficiently it is translated (Barbosa et al., 2013; Sonenberg and Hinnebusch, 2009). Protein stability and degradation processes further modulate the levels of functional protein in the cell (Ciechanover and Kwon, 2015; Sherman and Goldberg, 2001). Although studies have typically shown a positive correlation between mRNA and protein levels, the variability suggests that multiple mechanisms, including translation efficiency and protein stability, are significant in determining final protein levels in biological systems (Schwanhäusser et al., 2011; Liu et al., 2016). Several studies have reported average correlation coefficients around 40%–60%, indicating that mRNA levels indicate protein abundance but are far from perfectly predictive due to various biological and methodological confounders (Vogel and Marcotte, 2012; Perl et al., 2017; Kosti et al., 2016) (Figure 2). This makes studies involving the gene level resulting from bioinformatics analysis somewhat one-sided.
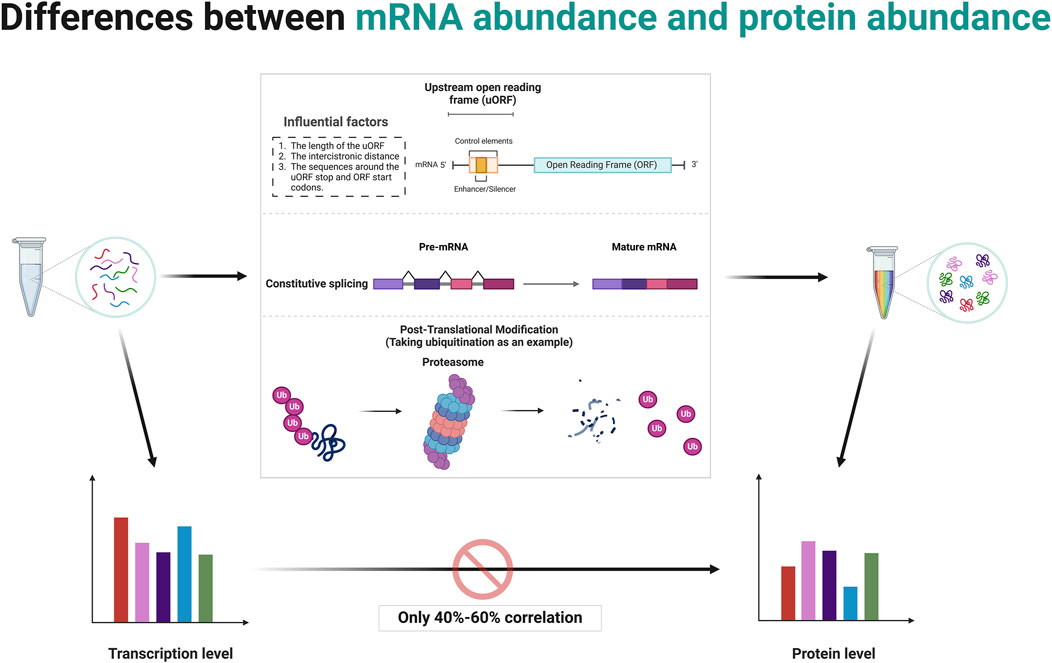
Figure 2. Post-transcriptional and post-translational modifications contribute to discrepancies between mRNA abundance and protein abundance. Created in BioRender. ZHAO, X. (2025) https://BioRender.com/q14t025.
3.1 The insights from the effect of upstream open reading framesThe concept of mRNA translation primarily involves coding mRNA into proteins by ribosomes, a process central to gene expression (Kozak, 1999; Jackson et al., 2010). Typically, this decoding focuses on the ORF that starts with a start codon (usually AUG) and ends with a stop codon (Jackson et al., 2010; Kozak, 2001). The discovery and study of uORFs have expanded our understanding of translational regulation and its complexities (Barbosa et al., 2013; Calvo et al., 2009; Wethmar, 2014). uORFs are alternative ORFs located upstream of the in the 5′untranslated region (5′UTR) of main coding sequence of an mRNA (Calvo et al., 2009). These uORFs can play a significant role in the regulation of translation of the main ORF (Barbosa et al., 2013; McGillivray et al., 2018).
uORFs are initiated when a ribosome recognizes and binds to a start codon (usually AUG, but sometimes a near-cognate codon) at the 5′UTR of an mRNA (Silva et al., 2019). The presence of a uORF upstream of the main coding sequence can alter ribosomal scanning and initiation dynamics, consequently affecting the translation of the downstream ORF (Hinnebusch et al., 2016; Morris and Geballe, 2000). After a uORF is translated, ribosomes can either dissociate from the mRNA or resume scanning for another start codon (Kozak, 2005). Several factors affect the ability of ribosomes to re-initiate translation at the downstream main ORF depends on, including the length of the uORF, the distance between the cistrons (the gap between the uORF and the main ORF), and the sequence context around the stop codon of the uORF and the start codon of the main ORF (Jackson et al., 2010; Ivanov et al., 2010; Kozak, 1987) (Figure 2). Often, efficient re-initiation is contingent upon the ribosomal retention of initiation factors during the uORF translation. The impact of a uORF on the main ORF translation can vary dramatically depending on its sequence and context (Pavitt, 2005). Some uORFs exhibit features that stall ribosomal function or slow translation, potentially enhancing or inhibiting the translation of the main ORF (Caliskan et al., 2015). For instance, Phan et al. elucidate how conserved uORFs in the 5′UTR of Polo-like kinase 4 (PLK4) mRNA play a crucial role in controlling the translation of PLK4, thereby regulating the duplication of centriole in primordial germ cells (PGCs) and preserving genomic integrity (Phan et al., 2022). This translational control mechanism prevents excessive PLK4 synthesis, vital for preventing centriole amplification and associated mitotic errors, highlighting a specific requirement for uORF in regulating the balance of PLK4 levels during germ cell development (Phan et al., 2022). A recent study by Cieśla et al. reveals that the regulation of SF3B1 protein levels through ALKBH5-driven N6-methyladenosine demethylation in the 5′UTR influences its translation, driving splicing mechanisms that impact DNA repair and epigenetic regulation (Cieśla et al., 2023). These studies demonstrate the critical role of post-transcriptional modifications in the expression of final protein.
3.2 The insights from alternative splicingAlternative splicing is a post-transcriptional regulatory mechanism that contributes significantly to proteomic diversity and gene expression regulation in eukaryotic organisms (Nilsen and Graveley, 2010; Wang et al., 2008; Black, 2003; Barash et al., 2010). It involves the selective inclusion or exclusion of pre-mRNA segments (exons) during the RNA splicing process, resulting in multiple distinct mRNA transcripts from a single gene (Chen and Manley, 2009; Kornblihtt et al., 2013) (Figure 2). This process can affect the quantity as well as the functionality of the encoded proteins (Kornblihtt et al., 2013). When determining RNA abundance using technologies like RNA-seq, reads mapping to a gene are typically aggregated to estimate the overall abundance of the gene (Wang et al., 2009; Mortazavi et al., 2008). This standard approach does not differentiate between the various transcripts produced by alternative splicing (Ozsolak and Milos, 2011; Trapnell et al., 2009). Consequently, even if the total mRNA of a gene remains constant, changes in the splicing patterns can lead to proteins with significantly altered types and functions (Black, 2003; Kalsotra and Cooper, 2011). This is a critical factor to consider in gene expression analysis because while the quantitative measure (total RNA transcripts) might not show variation, the qualitative changes (different splice variants) can have profound biological ramifications (Nilsen and Graveley, 2010; Barash et al., 2010). Meanwhile, specific conditions or stimuli might induce changes in splicing patterns without altering the overall mRNA levels (Wang et al., 2008; David and Manley, 2010). Such differential splicing events can produce protein variants with differing, sometimes opposing, functions (Wang et al., 2008; David and Manley, 2010). Some splice variants may include or exclude sequences with regulatory elements affecting translation efficiency, such as internal ribosome entry sites or uORFs (Barbosa et al., 2013; Sonenberg and Hinnebusch, 2009).
In a recent study, researchers demonstrated the significant role of alternative splicing in the regulation of chromatin dynamics, particularly through the manipulation of histone deacetylase (HDAC)7 splicing downstream of T cell signaling pathways (Agosto et al., 2023). Notably, the longer HDAC7 isoform, induced by the RNA-binding protein CUGBP Elav-like family member 2, enhances the expression of key T cell surface proteins such as CD3, CD28, and CD69, highlighting the broad implications of alternative splicing on histone modification and gene regulatory mechanisms in T cells (Agosto et al., 2023). Particularly in studies related to diseases such as cancer, where splicing patterns can be drastically altered, researchers must consider the total expression level of a gene as well as the expression levels of individual splice variants.
3.3 The insights from post-translational regulationDifferences in protein abundance and gene abundance are largely caused by post-translational modifications (PTMs). PTMs like ubiquitination and phosphorylation can target proteins for degradation, leading to lower protein levels despite high mRNA expression (Hershko and Ciechanover, 1998; Hunter, 2007; Cohen, 2000; Ciechanover, 2005; Deshaies and Ferrell, 2001). Conversely, protein levels rise when modifications protect proteins from degradation. PTMs also modulate protein activity, producing active or inactive forms that do not directly correlate with mRNA levels (Figure 2). For instance, phosphorylated proteins often exhibit functions or stabilities different from their non-phosphorylated counterparts (Hunter, 2007; Olsen et al., 2006; Manning et al., 2002). Modifications such as phosphorylation, methylation, and acetylation also impact protein-protein interactions, altering binding affinities and affecting signaling pathways and cellular processes independent of gene expression (Ross et al., 2023; Nishi et al., 2014; Duan and Walther, 2015). The regulatory mechanism of post-translational modifications is comprehensively summarized in Table 1.
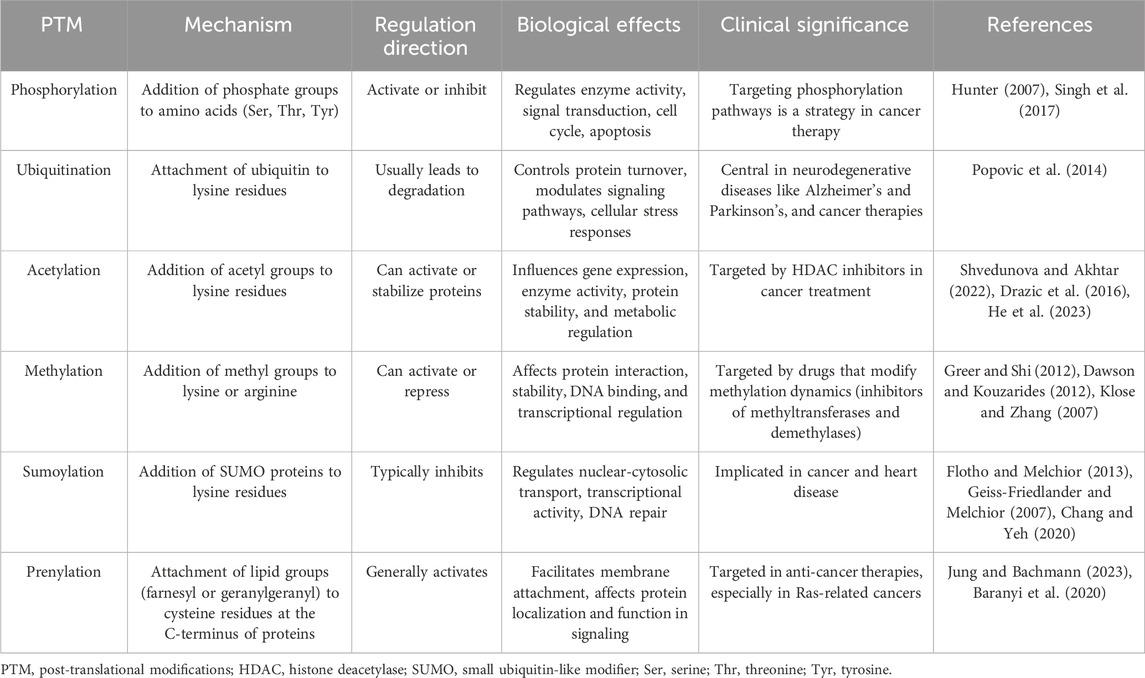
Table 1. Overview of post-translational modifications in protein regulation.
Integrative multi-omics analysis highlights the significant impact of PTMs on the differences in protein and gene levels, particularly during the human cell cycle (Parkes and Niranjan, 2019). While mRNA and translation data explain some variations in protein abundance, the remaining inconsistencies are primarily due to PTMs, which adjust protein levels post-synthesis (Parkes and Niranjan, 2019). In a study focusing on triple-negative breast cancer (TNBC), researchers identified tumor endothelial marker 8 (TEM8) as a key indicator of breast tumor-initiating cells (Xu et al., 2021). The study also highlighted the binding of estrogen receptor α to the promoter region of the ubiquitin E3 enzyme ankyrin repeat and SOCS box containing 10 (ASB10). It also activates ASB10 transcription, and ASB10 interacts with TEM8, thereby affecting the ubiquitination of TEM8 and ultimately affecting the TEM8 protein level (Xu et al., 2021). Thus, Indirect evidence indicates that variations in TEM8 mRNA and protein expression across BRCA subtypes may be attributed to post-translational modifications (Xu et al., 2021).
4 Regulation of tumor immune microenvironment by paradoxical genesThe process of TIME begins with the recognition of tumor-specific antigens by antigen-presenting cells, like dendritic cells, which capture and present these neoantigens to naïve T cells in lymph nodes, thereby initiating T-cell activation (Mellman et al., 2011). This activation triggers a series of immune responses, including the release of chemokines that attract more effector immune cells (such as CTLs, NK cells, and macrophages) to the tumor site, effectively infiltrating the tumor (Joyce and Fearon, 2015). Within the TIME, a pivotal change occurs as effector T cells reprogram immunosuppressive cells and alter the metabolic environment, diminishing the suppressive function of regulatory immune cells such as regulatory T cells (Tregs), myeloid-derived suppressor cells (Ho et al., 2015). This culminates in the direct cytotoxic attack on tumor cells by CTLs and NK cells, utilizing mechanisms like perforin and granzyme release to induce tumor cell apoptosis (Trapani and Smyth, 2002). Furthermore, some activated T cells differentiate into memory T cells, providing long-term surveillance and a rapid response mechanism against tumor recurrence (Sallusto et al., 2004) (Figure 3).
Recently, several scholars have dedicated their research efforts to understanding how paradoxical genes influence prognosis through the regulation of the TIME. Cao et al. analyzed data from TCGA and the gene expression omnibus, revealing that the expression of C-X-C motif chemokine ligand (CXCL)11 was elevated in colon cancer tissues compared to healthy tissues, and higher levels of CXCL11 correlated with improved survival outcomes (Cao et al., 2021). Furthermore, assessment of three independent datasets, including TCGA and two single-cell RNA sequencing datasets from Gene Expression Omnibus, in addition to immunohistochemistry data from a COAD patient cohort demonstrated that this tumor suppressor effect possibly due to its association with an increased presence of antitumor immune cells (CD8+ T cells, and CD56 NK cells), underscored CXCL11’s role in modulating the TIME (4). Furthermore, HERV-H LTR-associating 2 (HHLA2), a newly identified member of the B7 immune checkpoint family, is also a typical paradoxical gene (Yan et al., 2019). HHLA2 is minimally expressed in normal pancreatic tissues but shows significant upregulation from precancerous stages to invasive pancreatic ductal adenocarcinoma (PDAC), according to immunohistochemistry analyses on tissue microarrays (Yan et al., 2019). In 77.17% of PDAC cases, the expression of HHLA2 is strongly associated with an improved post-surgical prognosis, indicating functions of HHLA2 as a costimulatory ligand in pancreatic cancer, activating CD8+ T cell proliferation and improving patient prognosis (Yan et al., 2019). A subsequent study also presented a similar point; immunohistochemical analysis on tissue micro-arrays from surgically resected tumors of 122 pancreatic and 72 ampullary cancer patients revealed HHLA2 expression in 67% of pancreatic and 93% of ampullary tumors, associating enhanced expression with improved post-surgical outcomes, including delayed cancer recurrence and improved cancer-specific survival (Boor et al., 2020). Similarly, the study by Martinez-Turtos et al. highlights that overexpression of inositol-requiring enzyme 1α (IRE1α) in murine colorectal and Lewis lung carcinoma cells in syngeneic immunocompetent mice, leads to a tumor-suppressive phenotype (Martinez-Turtos et al., 2022). This anti-tumoral effect is attributed to the RNAse activity of IRE1α, which induces apoptosis in tumor cells, enhances adaptive anti-cancer immunosurveillance through XBP1 mRNA splicing, and regulates IRE1-dependent degradation of RNA (RIDD) (Martinez-Turtos et al., 2022). However, in addition to the tumor suppressive role of IRE1α, its tumor-promoting role is also evident in preclinical models of various cancers, including TNBC, PDAC, and colon cancer (Harnoss et al., 2020; Garcia-Carbonero et al., 2018; Xie et al., 2019). This duality suggests that the impact of Paradoxical genes on cancer prognosis may be multifaceted and not solely affected by the TIME (Figure 3).
5 Dual roles of paradoxical genes and associated signaling pathways in tumorsCertain genes and their associated signaling pathways exhibit bidirectional effects on tumor prognosis, transitioning between inhibiting and promoting tumor progression. This dualistic behavior is also one of the key factor contributing to the emergence of paradoxical genes (Hanahan and Weinberg, 2011; Sadikovic et al., 2008). This biphasic regulatory effect can depend on various factors, including tumor stage, tumor-specific expression, and TME (Hanahan and Weinberg, 2011; Plaks et al., 2015; Gerlinger et al., 2012; Marusyk et al., 2012; Bray, 2016). The impact of gene expression can vary by cancer type and the tissue of origin (Stratton et al., 2009). Genes beneficial in one type of cancer might be deleterious in another (Vogelstein and Kinzler, 2004; Hanahan and Weinberg, 2011; Sadikovic et al., 2008). The role of E-cadherin in tumor suppression is well-established in BRCA due to its function in maintaining cell-cell adhesion and inhibiting metastasis (Padmanaban et al., 2019). However, in other cases, such as gastric cancer, the expression of can be associated with different outcomes based on additional factors like the presence of specific mutations or the overall state of cellular adhesion molecules (Becker et al., 1994). We focuses on signaling pathways with biphasic regulatory effects, including the TGFβ, NOTCH, and NF-κB pathways (Figure 4). Genes associated with these pathways are key contributors to the development of paradoxical genes, which exhibit peculiar behaviors during gene expression and regulation.
6 Stage-specific expression of paradoxical genes: insights from the transforming growth factor-β signaling pathwaysThe expression of tumor suppressor genes exhibits significant variability across different cancer stages, contributing to the phenomenon of paradoxical genes (Sherr and McCormick, 2002). For instance, in the early stages of cancer, key genes like TP53 and RB1 play a crucial role in maintaining cellular integrity by regulating DNA damage repair and controlling cell cycle progression (Sherr and McCormick, 2002). However, as the tumor evolves, these genes often become inactivated due to mutations, deletions, or epigenetic modifications, leading to unchecked cell proliferation and advancement to more aggressive stages (Li et al., 1997; Cantley and Neel, 1999). PTEN, which regulates the PI3K/AKT signaling pathway, is commonly altered in cancers such as prostate and breast cancer, thereby facilitating tumor growth and survival (Li et al., 1997; Cantley and Neel, 1999; Salmena et al., 2008). In later stages, the suppression of tumor suppressor genes can precipitate metastasis and resistance to treatment, severely worsening the prognosis (Valastyan and Weinberg, 2011; Gottesman, 2002; Hanahan and Weinberg, 2000). Notably, the TGF-β pathway is recognized for its dual role in oncogenesis, acting as a tumor suppressor in initial stages while potentially fostering cancer progression in advanced stages (Massagué, 2008). Within this pathway, SMAD family members, including SMAD2 and SMAD4, are pivotal in relaying TGF-β signals that suppress cell division (Derynck and Zhang, 2003).
The TGFβ pathway suppresses tumors in the early stages of tumor development mainly by maintaining cellular homeostasis (including cell cycle arrest and apoptosis) and preventing uncontrolled cell proliferation (Colak and Ten Dijke, 2017). TGFβ regulates the cell cycle by inhibiting the transition from the G1 phase to the S phase, thereby preventing DNA replication and cell division, which is facilitated through the upregulation of cyclin-dependent kinase (CDK) inhibitors, which deactivate CDKs essential for cell cycle progression (Derynck, 1994; Decker et al., 2021). TGFβ also induces apoptosis, or programmed cell death through the activation of death-associated proteins and modulation of apoptosis-related genes, eliminating cells with potentially harmful mutations (Zhao et al., 2018; Schulte-Hermann et al., 1992). It also maintains cellular differentiation and proper morphology, thereby inhibiting the epithelial-mesenchymal transition (EMT), a critical process in cancer metastasis (Hao et al., 2019). Furthermore, TGFβ exerts anti-inflammatory effects within the cellular environment, regulating immune cell activity and cytokine production to suppress chronic inflammation, thus preventing tumor growth (Viel et al., 2016; Coussens and Werb, 2002).
In advanced cancer, TGF-β primarily acts as a tumor promoter. TGFβ promotes the EMT, a critical process for metastasis, by regulating transcription factors like Snail, Slug, and Twist that modify adhesion and migration properties of the cell (Peng et al., 2022; Wang et al., 2013; Ang et al., 2023). Simultaneously, TGF-β exerts systemic immune suppression and inhibits host immunosurveillance and also regulates the infiltration of inflammatory/immune cells and cancer-associated fibroblasts in the TME, causing direct changes in tumor cells (Yang et al., 2010). Neutralizing TGF-β enhances CD8+ T-cell- and NK-cell-mediated anti-tumor immune responses and increases the neutrophil-attracting chemokine production, leading to the recruitment and activation of neutrophils with an antitumor phenotype (Yang et al., 2010). It also interacts with cancer-associated fibroblasts and mesenchymal stem cells within the TME to remodel the extracellular matrix, increasing tumor stiffness and spreading cancer (Arima et al., 2023) (Figure 4).
The development of inhibitors that target TGFβ signaling is a promising treatment approach for cancers where TGFβ promotes tumor growth and metastasis (Derynck and Akhurst, 2007; Herbertz et al., 2015). These inhibitors generally block TGFβ receptors, preventing the downstream signaling cascades that lead to oncogenic effects (Herbertz et al., 2015). SB-431542 was initially identified as a potent and specific inhibitor of the activin receptor-like kinase (ALK)4, ALK5, and ALK7 type I receptors of the TGF-β superfamily, effectively and selectively inhibiting activin and TGF-β signaling without impacting BMP signaling or other divergent pathways like extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), or p38 mitogen activated protein kinase (Inman et al., 2002). In recent years, galunisertib (LY2157299 monohydrate), a selective, small molecule that can be taken orally to inhibit TGF-β receptor I kinase, exhibits antitumor activity across various cancer models, including breast, colon, lung, and HCC, by specifically downregulating SMAD2 phosphorylation and inhibiting the canonical TGF-β pathway (Herbertz et al., 2015). This drug is currently being evaluated in clinical trials in an intermittent dosing regimen (14 days on/14 days off, on a 28-day cycle) as part of monotherapy or in combination with other antitumor treatments to balance efficacy and safety, targeting cancers with high unmet medical needs like glioblastoma, pancreatic cancer, and HCC (Herbertz et al., 2015; Akhurst and Hata, 2012; Nadal et al., 2023). Clinical trials investigating combinations of TGFβ inhibitors with programmed cell death protein 1(PD-1)/programmed death-ligand 1(PD-L1) inhibitors are exploring this approach, showing promising results in improving anti-tumor immunity and patient outcomes (Wrzesinski et al., 2007). Hence, understanding the tumor stage and the specific role of TGFβ is crucial to determining when and how to target this pathway effectively. Personalizing treatment based on the genetic and molecular profiles of individual tumors could optimize the efficacy of TGFβ inhibitors and minimize adverse effects.
7 Tumor-specific expression of paradoxical genes: insights from the NOTCH signaling pathwayThe NOTCH signaling pathway is a crucial cell communication mechanism that influences various biological processes, such as differentiation, proliferation, apoptosis, and stem cell maintenance (Bray, 2016). This pathway involves NOTCH receptors (NOTCH1-4) interacting with delta-like (DLL1, DLL3, DLL4) or Jagged (JAG1, JAG2) ligands on adjacent cells, initiating proteolytic cleavages that release the NOTCH intracellular domain (NICD) (Bray, 2016). This domain moves to the nucleus, where it converts recombination signal binding protein for immunoglobulin kappa J region from a repressor to an activator, with mastermind-like proteins, initiating transcription of target genes families like HES and HEY (Bray, 2016). The finely tuned regulation of this pathway, which includes endocytic trafficking and post-translational modifications, is essential for maintaining cellular and tissue homeostasis (Fortini, 2009). The dual nature of NOTCH signalling in cancer biology—acting as a tumor suppressor in some contexts while promoting tumor progression in others—underscores the complexity of its signaling pathways and their diverse effects on cancer etiology and progression (Valdez and Xin, 2013) (Figure 4).
In specific cancers like those of skin and liver, NOTCH signaling is essential for maintaining cellular differentiation and tissue architecture (Panelos and Massi, 2009; Kawaguchi and Kaneko, 2021). Specifically, in squamous cell carcinoma, increased NOTCH signaling is associated with reduced tumor formation and progression (Panelos et al., 2008). Impaired NOTCH signaling, as demonstrated by the expression of the pan-NOTCH inhibitor dominant negative mastermind-like (DNMAML)1 in conditional transgenic mice, leads to hyperplastic epidermis and spontaneous development of cutaneous squamous cell carcinoma (SCC) and actinic keratoses, suggesting a protective role of canonical NOTCH signaling against cutaneous SCC (Proweller et al., 2006). Meanwhile, NOTCH1 signaling significantly inhibits the growth of HCC by inducing cell cycle arrest at the G (0)/G (1) phase and promoting apoptosis, by downregulating key cell cycle proteins and upregulating p21 and p53, while also suppressing antiapoptotic B-cell lymphoma 2(Bcl-2) expression (Giovannini et al., 2016; Giovannini et al., 2014; Kim et al., 2017; Qi et al., 2003; Viatour et al., 2011). However, the role of NOTCH signaling is paradoxically reversed in other types of cancers. In T-cell acute lymphoblastic leukemia (T-ALL) and certain breast cancers, NOTCH activation enhances cell proliferation, survival, and stemness, thereby promoting tumor growth and survival (Weng et al., 2004; Reedijk et al., 2005). Besides, the NOTCH signaling pathway also presents a biological dual nature widely in various cancers (Table 2).
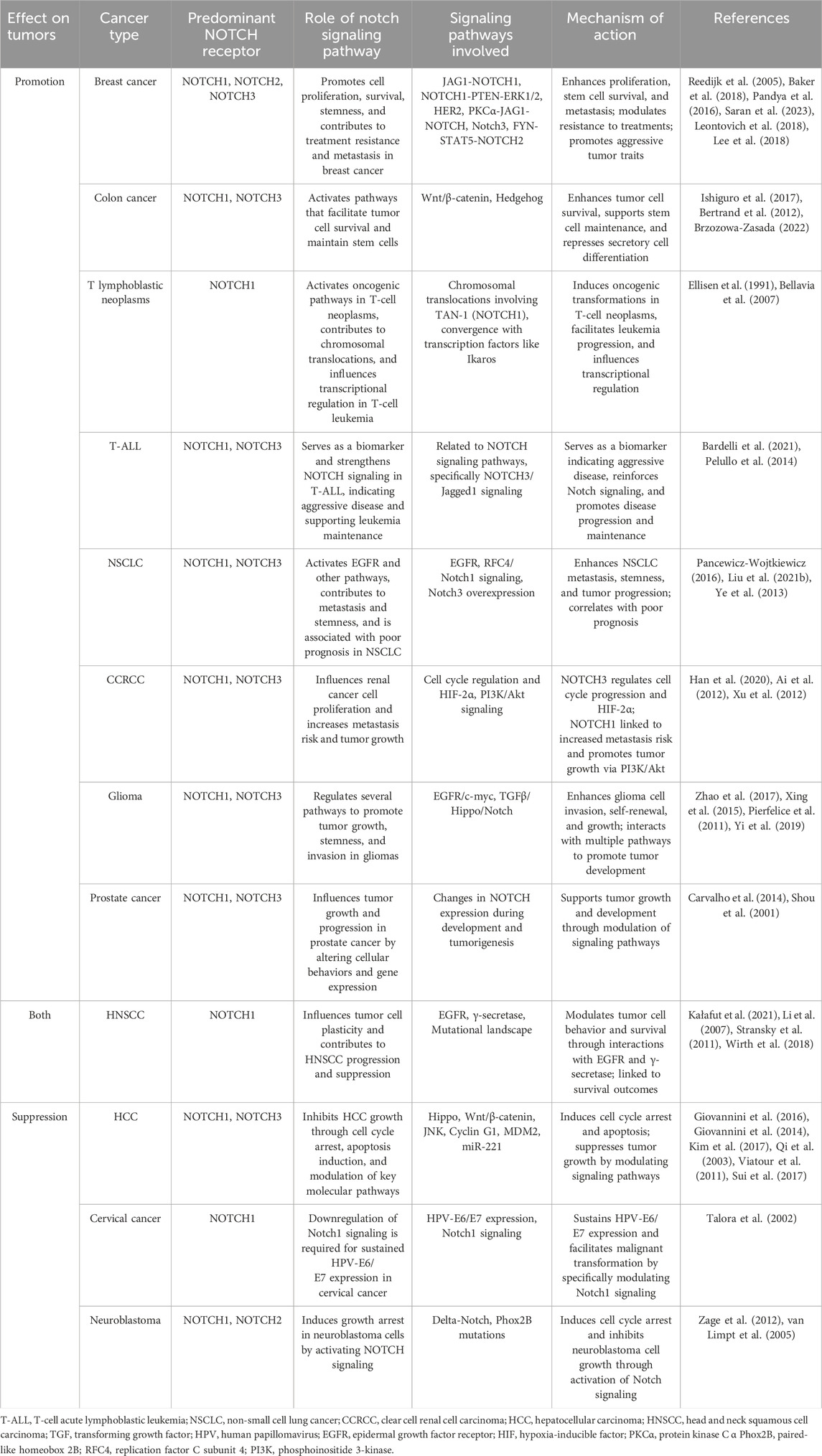
Table 2. Summary of the tumor-promoting or tumor-inhibiting effects of the NOTCH signaling pathway in various cancers.
8 The impact of environmental sensitivity on the formation of paradoxical genesThe environmental sensitivity of suppressor genes refers to the fact that the expression and function of these genes are affected by the TME, including factors such as hypoxia and acidity, which in turn affects cancer progression and cellular behavior (Gatenby and Gillies, 2004; Webb et al., 2011). Additionally, immune cells within this microenvironment release a variety of cytokines and growth factors that significantly impact cancer dynamics (Coussens and Werb, 2002; Grivennikov et al., 2010; de Visser et al., 2006). Specifically, certain immune-derived factors may suppress tumor cells, while others might activate signaling pathways that induce tumor cells (Coussens and Werb, 2002; Grivennikov et al., 2010; de Visser et al., 2006). These regulatory effects of environmental sensitivity on tumor suppressor genes undoubtedly contributed to the emergence of paradoxical genes.
8.1 Insights from hypoxiaHypoxia within the TME critically influences the progression of cancer by modulating tumor suppressor genes, primarily via the stabilization of hypoxia-inducible factors (HIFs) such as HIF-1α and HIF-2α (Semenza, 2003; Keith et al., 2011). Under low oxygen conditions, these transcription factors translocate to the nucleus, activating genes that drive angiogenesis, metabolism, cell survival, and invasion (Semenza, 2003). The suppression of the VHL gene under hypoxic conditions leads to unregulated HIF activity, promoting the secretion of angiogenic factors like vascular endothelial growth factor and platelet-derived growth factor, which are instrumental in tumor growth and proliferation (Semenza, 2003). Hypoxia plays a significant role in the regulation of tumor suppressor genes. For instance, Chen et al. discovered that Hypoxia-inducible HIF-1α directly interacts with Mdm2, enhancing the in vivo association between p53 and HIF-1 alpha and acting as a mediator in their indirect interaction, which is crucial for the stabilization and activation of p53 in response to hypoxic stress (Chen et al., 2003). Furthermore, they found that HIF-1 alpha inhibits the Mdm2-mediated ubiquitination and nuclear export of p53, thereby protecting p53 from degradation and facilitating its role in transcriptional activation under hypoxic conditions (Chen et al., 2003). Additionally, PTEN, a crucial regulator of the PI3K/AKT signaling pathway, is downregulated by microRNAs like miR-21, which are themselves upregulated under hypoxic conditions. This indicates that hypoxia indirectly plays a significant role in the regulation of PTEN through the modulation of microRNA levels (Cascio et al., 2010; Kulshreshtha et al., 2007; Meng et al., 2007).
Hypoxic stress within solid tumors profoundly influences epigenetic regulation, particularly affecting DNA methylation and histone modifications (Shahrzad et al., 2007; Krieg et al., 2010). For instance, Watson et al. investigated the effects of chronic hypoxia in prostate cells and identified significant epigenetic changes, including increased global DNA methylation and H3K9 histone acetylation, associated with an altered cellular phenotype characterized by enhanced apoptotic resistance, cellular senescence, and increased invasiveness (Watson et al., 2009). These findings suggest that chronic hypoxia induces genome-wide adjustments in DNA methylation and histone modifications, potentially promoting and maintaining a hypoxic-adapted cellular phenotype that may contribute to tumor development (Watson et al., 2009). Meanwhile, Krieg et al. demonstrated that HIF-1α regulates the histone demethylase JMJD1A, which enhances the expression of hypoxia-responsive genes and promotes tumor growth (Krieg et al., 2010). This study highlights the critical role of HIF-1α in modifying histone enzymes under hypoxic conditions, thereby contributing to the dynamic regulation of gene expression in cancer cells (Krieg et al., 2010). These epigenetic modifications could further facilitate the emergence of paradoxical genes by modulating the expression diversity of tumor suppressor genes.
These molecular alterations under hypoxic stress lead to severe consequences, including enhanced angiogenesis, facilitating metastatic spread, altered cellular metabolism favoring cancer cell survival in low oxygen conditions, and increased resistance to conventional therapies (Semenza, 2003; Harris, 2002; Semenza, 2010). Targeting these adaptations has led to novel therapeutic strategies aimed at the hypoxic niche—such as the development of inhibitors that block HIFs, strategies to restore the functions of inactivated tumor suppressor pathways, and the use of hypoxia-activated prodrugs (Semenza, 2003; Wilson and Hay, 2011; Brown and Wilson, 2004; Wigerup et al., 2016). Additionally, addressing the downstream effects of tumor suppressor gene suppression, such as the use of PI3K inhibitors in cases of PTEN loss, offers a refined approach to disrupt the survival mechanisms employed by tumor cells in the hypoxic TME (Sansal and Sellers, 2004; Janku et al., 2018). These approaches leverage the modulation of tumor suppressor genes within the hypoxic tumor microenvironment to enhance the efficacy of cancer treatment.
8.2 Insights from acidosisAcidosis within the TME significantly also impacts the cancer cell behavior (Webb et al., 2011; Damaghi et al., 2013). This condition stems from metabolic alterations in tumor cells, notably the high rates of glycolysis leading to excessive lactic acid production, even in the presence of oxygen (Webb et al., 2011; Gillies et al., 2008). This metabolic shift results in an accumulation of lactic acid, leading to a marked decrease in pH within the surrounding tissue (Gillies and Gatenby, 2007). Meanwhile, Riemann et al. demonstrated that an acidic tumor microenvironment induces reactive oxygen species (ROS) formation which then activate mitogen-activated protein kinases (MAPK) signaling in cancer cells (Riemann et al., 2011). This activation leads to phosphorylation of the transcription factor CREB via p38, altering transcriptional activity and potentially sustaining tumorigenic changes even after cells return to a normal environment (Riemann et al., 2011). Additionally, the acidic environment can further lead to epigenetic changes, affecting DNA methylation and histone modifications, potentially leading to the silencing of tumor suppressor genes or the activation of oncogenes (Thorne et al., 2009; Kulis and Esteller, 2010). These insights highlight the regulatory influence of pH factors on tumor prognosis and related signaling pathways, thereby creating conditions that are favorable for the emergence of paradoxical genes.
8.3 Insights from immune signaling pathways modulationImmune modulation can have profound effects on the expression and function of tumor suppressor genes (Oeckinghaus et al., 2011). A key player in this regulatory network is the NF-κB signaling pathway, which orchestrates responses that can either inhibit or promote tumor progression based on the surrounding cellular context (Karin et al., 2002; Baud and Karin, 2009). NF-κB promotes oncogenesis by upregulating the expression of genes that promote cell proliferation and inhibit programmed cell death. Key targets include genes encoding anti-apoptotic proteins, such as Bcl-2, B-cell lymphoma-extra large (Bcl-xL), and inhibitor of apoptosis proteins, cell cycle regulators (such as cyclin D1 and c-Myc), and growth factors that together foster an environment conducive to the survival and proliferation of cancer cells (Annunziata et al., 2007; Karin et al., 2002; Baud and Karin, 2009). This role of NF-κB has been extensively documented in cancers like multiple myeloma, where it contributes directly to the survival and proliferation of malignant cells under chemotherapeutic stress (Annunziata et al., 2007). Additionally, NF-κB is a critical regulator of the TME by stimulating the production of pro-inflammatory cytokines and chemokines such as TNF-α, interleukin (IL)-6, and IL-8. These molecules aid in reshaping the surrounding stroma, promoting angiogenesis, and facilitating tumor cell invasion and metastasis (Karin, 2006). Furthermore, NF-κB helps recruit and activate various immune cells within the TME that support tumor growth rather than combat it, thus contributing to tumor progression and the suppression of effective anti-tumor immune responses (Karin, 2006). NF-κB also contributes to the ability of tumor cells to evade immune surveillance. It modulates the expression of molecules affecting the immune response, such as major histocompatibility complex molecules and PD-L1, a ligand for the PD-1 receptor on T cells, which inhibits T cell function. NF-κB promotes an immunosuppressive microenvironment by enhancing the expression of PD-L1 on tumor cells, allowing tumor cells to escape detection and destruction by the immune system (Greten and Karin, 2004).
The NF-κB also plays key role as a suppressor of tumor development under certain contexts (Perkins, 2012). This suppression is primarily evident during the early stages of cancer and involves mechanisms that maintain cellular homeostasis and inhibit malignant transformations (Perkins, 2012). NF-κB contributes to genomic stability maintenance by regulating the expression of genes involved in DNA repair and cell cycle checkpoints. This function prevents the accumulation of genetic mutations that could otherwise lead to oncogenesis (Volcic et al., 2012). NF-κB can also induce cellular senescence, a permanent cell cycle arrest that functions as a barrier against the proliferation of potentially cancerous cells. This dual role of promoting DNA repair and senescence helps to suppress early tumor development and progression (Janssens and Tschopp, 2006). In specific cellular contexts, NF-κB can activate the transcription of certain tumor suppressor genes. For example, NF-κB induces the expression of GADD45β, a stress-response gene that plays a crucial role in DNA repair and cell cycle regulation (Jarome et al., 2015; Al Tarrass et al., 2024; De Smaele et al., 2001). By activating such genes, NF-κB contributes to the activation of mechanisms that can curb uncontrolled cell growth and promote apoptotic pathways in cells that have undergone malignant transformation (De Smaele et al., 2001) (Figure 4).
The ubiquitous role of the NF-κB signaling pathway in cancer makes it a significant target for therapeutic intervention (Karin, 2006). Current ther
留言 (0)