Eligible patients: all inpatient children with (a) simple orthopedic percutaneous metal removement; (b) dressing/change/debridement/wound flushing; or (c) other minor surgical intervention between the ages of 6 and 15 years, who present to the outpatient clinic of the Clinic of Pediatric Surgery at Inselspital University Hospital, Bern, Switzerland.
Inclusion criteria: (a) indication to undergo an elective minor surgical procedure (removal of percutaneous osteosynthesis or pin material or dressing change or other minor surgical intervention) and (b) age minimum of 6 years, maximum of 15 years.
Exclusion criteria: (a) inability to understand the VR program; (b) inability to complete the questionnaire due to language deficiencies; (c) neurologic disorders; (d) respiratory tract infections; and (e) intolerance of the VR headset or VR gaming procedure.
There are no prohibited concomitant interventions during the trial, as it has never been necessary to prohibit any interventions or medications when using the standard medication Kalinox.
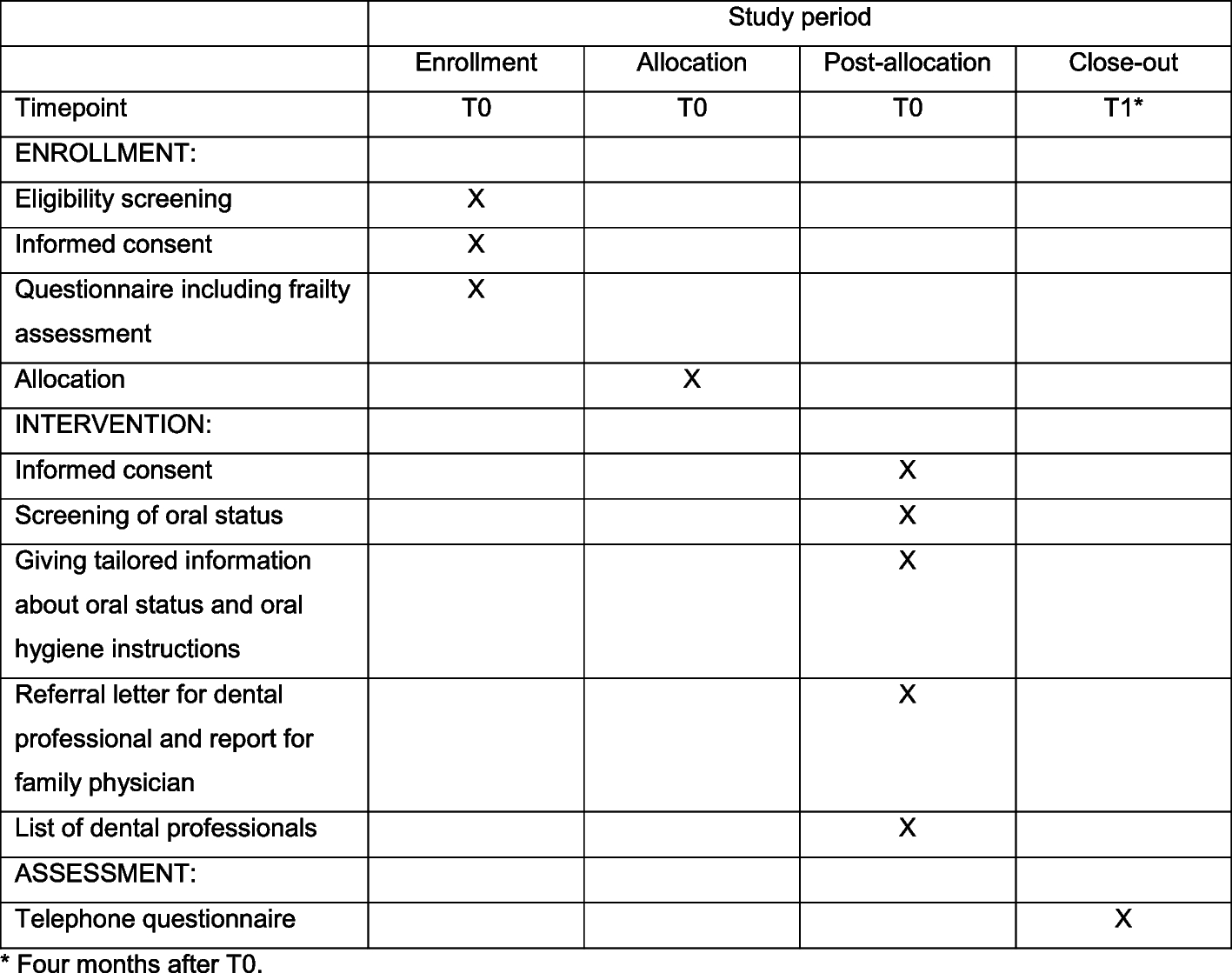
Endpoints and safety objectivesIntroducing remarks
Selecting the endpoints (outcome measures) is crucial for the design of the study. The outcome measure should be responsive, valid, and short. In addition, the outcome should be self-reported, thereby eliminating inter-observer bias and easing the administration burden. The study results should aim to assess long-term safety of VR application in children for analgesia and distraction and its tolerability. Short questionnaires are known to be less of a burden in clinical trials [25] and result in improved patient compliance and response rates, and are thought to improve the quality of the patient’s response [26]. For example, for a commonly used score in orthopedics, an item reduction from 17 to 8 led to a reduction of incomplete data from 24 to 6%, thereby strengthening the data quality and the appeal of the shorter version [27]. To assess validity, a gold standard would be necessary, which unfortunately does not exist. As an alternative, current pain ratings (cognitive, affective, and sensory) on a graphic rating scale (GRS) ranging from 0 to 10 are commonly used as an outcome measure. Of note, it is not the aim of the study to assess if the patients immerse into the VR environment. However, we plan to assess the amount of immersion due to VR/analgesia by nitrous oxide by the patient’s reaction to light touch and the patient’s reaction to passive lifting of one of the extremities. Available specific outcome measures in the literature for the assessment of pain levels: there are numerous scores to measure pain levels in different ages of children in the literature. We want to measure the pain intensity with the visual face scale of Bieri or with a visual analog scale (VAS 0–100).
Of these, the following are validated for the use in children:
1.
Likert scale for pain intensity.
2.
Questionnaire including fun level, motivation for replication.
3.
Clinical parameters like heart frequencies during the procedure, and blood pressure before and after intervention.
Satisfaction at the end of the therapy/procedure.
4.
Questionnaires
Use of these available outcome scores in targeted medical journals
When searching the PubMed literature database regarding the use of these scores in the targeted journals for this study in 2018, we could identify the following review study of 34 studies and most often used was the VAS (n = 19), followed by the face scale (n = 8) and other graphic scales (n = 21). These results clearly show that the outcome score VAS and face scale are age dependent and currently the preferred technique in the literature.
Disease-specific primary outcome measures
Given the information above, we have selected the VAS for children and adolescents aged 10–15 years and visual face scale of Bieri for children aged 6–9 years as the primary outcome measure for pain in our study. These have been reported to be the most commonly used outcome measure: In a review evaluating the utility of VR as a distraction method to alleviate pain and distress during medical procedures, most of the reviewed studies used the same outcome measure that we have planned [18].
In addition to the patient-reported pain scale, vital parameters including heart rate frequency and blood pressure will be measured before, during, and after the procedure to add to the measure of a potential pain reaction.
Disease-specific secondary outcome measure
The application of Kalinox® is used in our clinic in children with a time limit of 20 min. In our study, we would like to investigate whether VR distraction also has a time limit compared to the application of Kalinox® by assessing the amount of time during which VR is applied.
Non-disease-specific secondary outcome measures
As there is no “gold-standard” for the assessment of anxiety in pediatric patients, it appears worthwhile to also assess other, non-disease-specific outcome measures. Fun is generally not associated with a medical intervention; however, we will assess fun as an outcome measure, as described in the patient assessment form.
Assessment of patient satisfaction is a further endpoint with treatment outcome, as this is the ultimate goal against which all other scores have to be compared. Since even the assessment of patient satisfaction is not standardized, we chose a system that our group has experience with. In our system, the patient is asked if he/she would undergo the same procedure again.
Role and responsibilities of the study execution
The study is based in the pediatric clinics of the University Hospital Bern, Inselspital, and registered in the study registry of the University Hospital Bern. It is supervised by the research coordinator of the Children and Adolescents Medicine Division of the Inselspital, University Hospital Bern. The research coordinator and the University Hospital's DLF study portal provide support in day-to-day matters and share information about other committees and important information. The exchange takes place via the PI as the point of contact.
The recruiting physician or the doctoral student is responsible for screening hospitalized patients and providing these patients with detailed information about the study. If the patients are willing to participate, the recruiting physician or the doctoral student coordinates with the regular patient management team to schedule the study appointment, ensuring the appropriate time slot is allocated for the study. The recruiting physician or the doctoral student is also involved in overseeing the overall progress of the study and ensures all necessary documentation is in place.
The study nurse works closely with the outpatient clinic staff, supporting the daily clinical activities and assisting with study-related tasks. This includes the collection of informed consent forms and the administration of study procedures. The study nurse ensures that all study-related activities are carried out in accordance with the protocol.
On a monthly basis, the recruiting physician or the doctoral student, study nurse, and PI meet to review the progress of the study. During these meetings, they discuss the implementation of the study, address any challenges, and ensure the study is being conducted in compliance with the protocol.
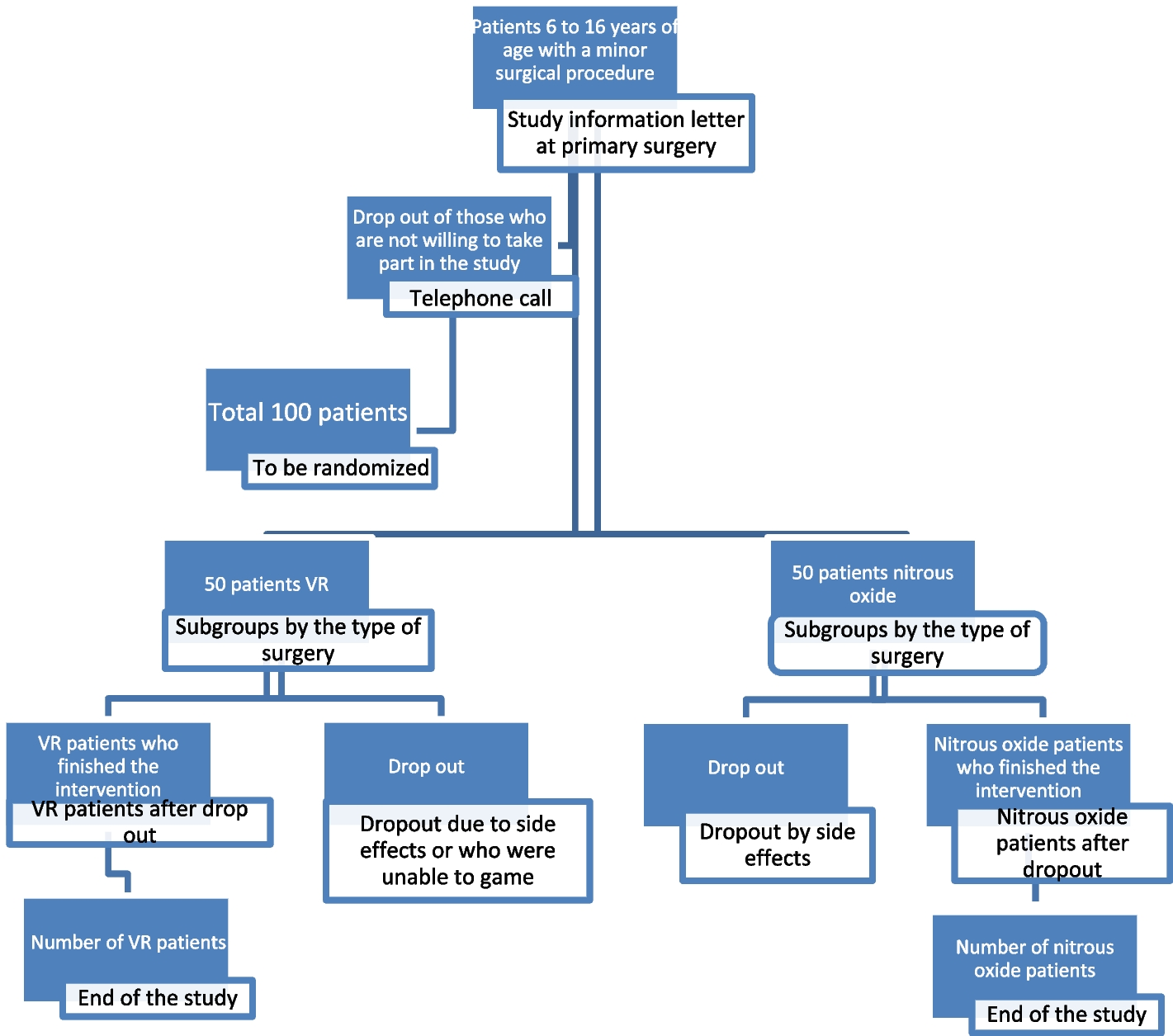
Mode of recruitment, intervention, and administration of the questionnaires
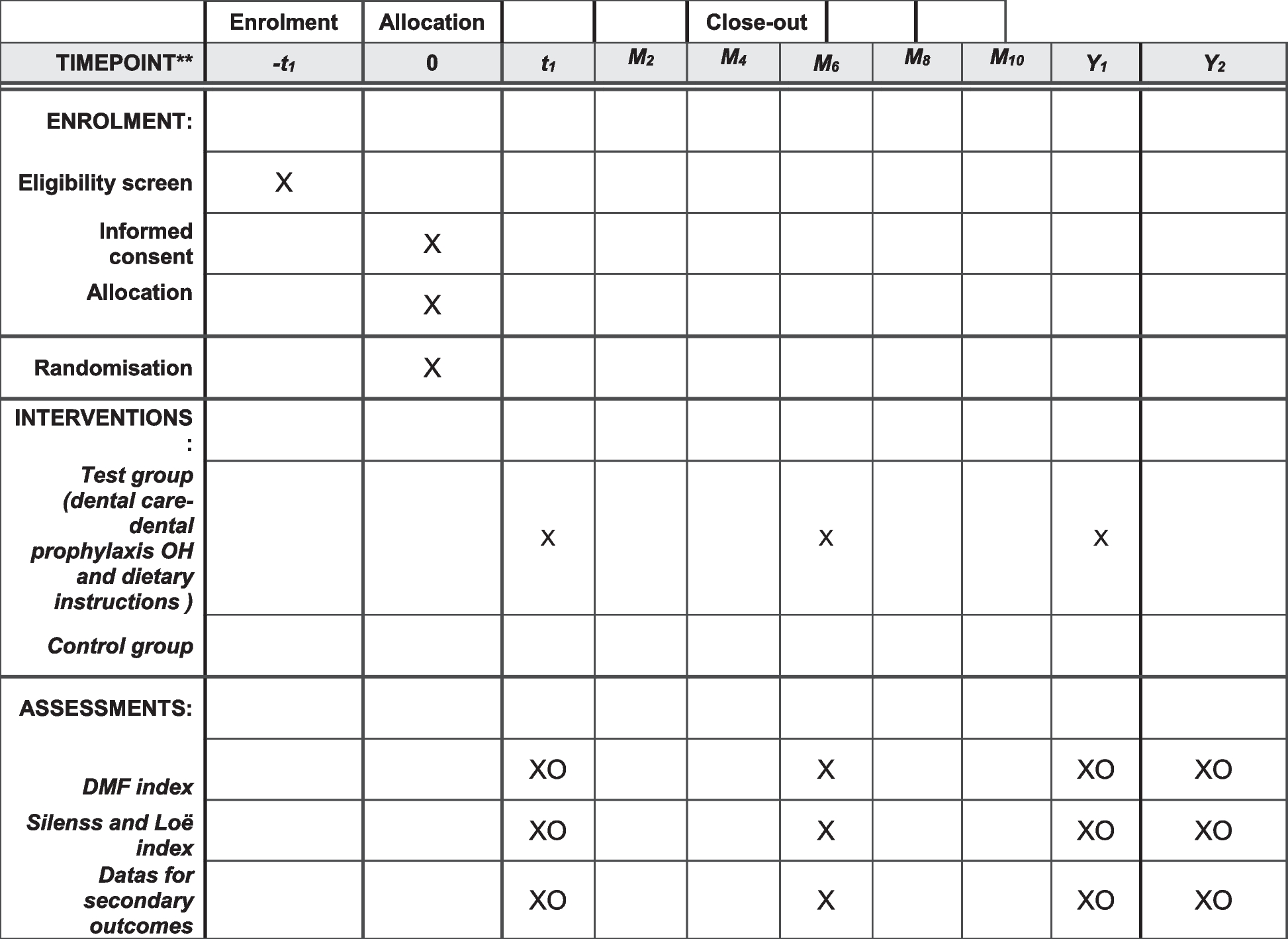
During the hospital stay of the primary surgery, we verbally inform the patients who fulfil the eligibility criteria and their parents about our study and we inform them in writing with the study information sheet, as attached. The information also includes an informed consent form. One to 2 weeks thereafter, the family will be contacted by a phone call if they agreed to be planned for a study slot in our outpatient clinic. On the day of the surgical procedure in our outpatient clinic, the informed consent will be collected and checked that it has been signed by all parties (adolescents, parents, and investigator). A web-based randomization will be done for those with a positive reply. The patient will be informed if he/she has been allocated to the VR or the control group (nitrous oxide). VR gaming in detail will be explained, if necessary, by the introduction modus of the game “Eagle Flight” and the trial intervention is completed as described above (see chapter trial intervention). During the procedure, a clinic-internal Kalinox® evaluation form is employed recording the amount of time, additional administered medication, and occurring side effects. We similarly use this form in the VR group and the control group to document the administration. In addition, vital parameters are measured before, during, and after the surgery. The questionnaire (see additional file 5) which assesses pain, patient satisfaction, and fun during the treatment will be filled out by the patient 15 min after the intervention. Two weeks after the procedure, the patients will be asked by mail to complete the same questionnaire again to avoid possible biased answers under the influence of the intervention or nitrous oxide application. Non-responding participants are reminded three times by mail; at the second timepoint, a letter is sent together with the questionnaire. Participants still not responding are contacted by phone to determine the reason for non-responding. In cases where patients or their parents withdraw their informed consent, collected data will still be used for further analysis and will be stored in an encrypted pattern. If possible, an invitation by mail will be sent to patients still not responding.
Randomization
All participants have an equal probability of assignment to the groups (1:1 allocation). External randomization is achieved by means of computer-generated random number tables in blocks of variable number [2,3,4] of subjects, stratified by indication (a: orthopedic metal removal; b: dressing/change/debridement/wound flushing; and c: other minor surgical interventions). We also stratified the groups by age (a: 6–9 years and b: 10–15 years) to reduce a possible confounder of age. Considering that the primary outcome, pain, is otherwise identical in both age groups and since it is known from the daily practice of anesthesiology that our different assessment tools for the two age groups (visual face scale of bieri and VAS) provide identical results regarding the intensity of pain, we would like to evaluate these two groups together.
These allocation tables are created in RedCAP (Research Electronic Data Capture) [28,29,30]. The randomization tables are stored at the CTU (clinical trials unit), and the authors and the treating physicians do not have access to these lists.
At the enrollment of participants, the recruiting physician/the study nurses or our master or doctoral student(s) register the patient in a web-based database. After registration, the patients are informed about the randomization result. The information, as to which group the patient has been randomized to, is forwarded to the nurses who take care of the administration of the VR program or nitrous oxide.
The method of generation of the random number tables, especially the block size, is unknown to the treating physicians.
Benefit/risk relationship for the patient
Since we assume that the VR program will result in decreased anxiety and pain levels, we believe that the risk for the patient is not greater than not participating in the study at all. However, if the VR program results in decreased anxiety and pain, there is a potential benefit for the patient.
If VR gaming has to be stopped early when the patient does not tolerate the VR application or when the pain management is insufficient, we will apply rescue medication and can always change to the standard procedure, using nitrous oxide.
In summary, there is no risk for the patient; however, a possible benefit might emerge. According to possible known side effects, the review of Scapin et al. [6] described in 34 analyzed studies that there are no major side effects when VR technology is employed.
Patients age according to the principle of subsidiarity
We decided to test children across all age groups, as in our clinical experience, younger children enjoy the entertainment with video clips as much as older children. Especially in the younger age group, nitrous oxide application is sometimes not possible due to more anxiety to the inhalation mask. Therefore, the younger age group is rather experienced with video clips than with inhalation masks. As such, we believe there is more of a benefit than a risk in these children.
Side effects
With the VR application, it has been reported that some children complain of nausea during active gaming [6]. This side effect ceased directly after ending the gaming activity. In our standard medication with nitrous oxide, side effects such as nausea, vomiting, dizziness, headache, mood changes, hyperventilation, and headache can always occur. We will record these mentioned expected side effects in a structured manner for every participant. Overall, side effects should be reduced in our study, due to the less frequent side effects in the VR group [31].
For other unexpected adverse events, the investigator and sponsor conduct a causality assessment of the event to the trial intervention and a severity assessment of the event as mild, moderate, or severe. Any event assessed as possibly, probably, or definitely related is classified as related to the trial intervention. Mild means the complication is tolerable, moderate means it interferes with daily activities, and severe means it renders daily activities impossible [24].
Serious adverse event (SAE)
1.
Results in death or is life-threatening
2.
Requires in-patient hospitalization or the prolongation of existing hospitalization,
3.
Results in persistent or significant disability or incapacity, or
4.
Causes a congenital anomaly or birth defect
In our study, SAE could occur because of nitrous oxide application, if the young patient is pregnant, either known or unknown. An allergic shock reaction even could arise to any given medication, nitrous oxide, NSAR in advance, or intranasally applied fentanyl. A fall from the stretcher with additional trauma due to the fall is not to be expected in the VR group, due to the protection of the assisting nurse.
Reporting of SAEs
All SAEs are documented and reported immediately (within a maximum of 24 h) to the sponsor–investigator of the study. If it cannot be excluded that the SAE is attributable to the intervention under investigation, the investigator reports it to the Ethics Committee via BASEC within 15 days. No SAE that could be related to the investigational device or study procedures are expected during this clinical trial. Such adverse effects are recorded and reported according to the recommendation given in the extension of the CONSORT Statement “Better Reporting of Harms in Randomized Trials” [32].
Data managementHardware and software
The case report forms (CRFs) in this trial are implemented electronically using a dedicated electronic data capturing (EDC) system (REDCap). The EDC system is activated for the trial only after successfully passing a formal test procedure. All data entered in the CRFs are stored on a Windows server in a dedicated mySQL database.
Responsibility for hosting the EDC system and the database is CTU Bern.
Data security, access, and backup
Data security is handed over to the CTU of the University of Bern. The server hosting the EDC system and the database is kept in a locked server room. Only the system administrators have direct access to the server and backup tapes. Personal passwords (site investigator, statistician, monitor, administrator, etc.) regulate permission for each user to use the system and database as he/she requires. All data entered into the CRFs are transferred to the database using Secure Sockets Layer (SSL) encryption. Each data point has attributes attached to it identifying the user who entered it with the exact time and date. Retrospective alterations of data in the database are recorded in an audit table. Time, table, data field and altered value, and the person are recorded (audit trail). A multi-level backup system is implemented. Backups of the whole system including the database are run internally several times.
Access to these data is password protected and is limited. In case of inspections by the Ethics Commission, access is granted to the raw data. The master key table, which allows the linkage of patient data to study data is locked.
Analysis and archiving
At final analyses, data files will be extracted from the database into statistical packages to be analyzed. The status of the database at this time is recorded in special archive tables. The study database with all archive tables will be securely stored by CTU Bern for at least 15 years. The sponsor also keeps the trial master file and final reports both in electronic and in hard copy form for at least 10 years.
Confidentiality
To maintain participant confidentiality every participant is given an identification number, which is stored in a Master key table separately from the study data. It is the only file, which allows the linkage of patients’ data to study data and will be locked and stored separately from study data.
Statistical significance and clinical importance
Once the study results are available, differences in the outcome between treatment groups will be analyzed. To compare these differences, various parametrical or non-parametrical statistical tests are used which result in probability values that are compared to predefined levels of significance. However, it has been demonstrated that statistical significance alone does not necessarily denote clinical relevance [33]. Sometimes, statistically significant results may not be clinically important. Insignificant results do not rule out the possibility of clinically important effects [33]. For that reason, the concept of the “Minimal Clinically Important Difference” (MCID) has been developed. It is defined as the “smallest treatment effect that would result in a change in patient management, given its side effects, costs and inconveniences.” [33]. However, in pediatrics, it has mainly been used for the evaluation of treatment outcomes in rheumatic diseases [34] and less for the evaluation of anxiety.
Power calculations and sample size
Since the aim of the study is to demonstrate that the treatment in the VR group is not inferior (i.e., equivalent or possibly superior) than the nitrous oxide treatment, this study has to be categorized as a non-inferiority study. Unfortunately, there are no comparable studies up to this time, that have assessed the preoperative anxiety in children, so the treatment effect sizes are unknown. As such, a power calculation is not possible. Therefore, we have agreed to recruit 50 patients in each treatment group, resulting in 100 children altogether. The absence of a power analysis will be respected in the discussion of the study results, where we will report our intended sample size. This assures transparency to the reader and is necessary to assess, if the power of the trial was sufficient to detect a possible existing treatment effect.
Baseline inquiry
The questionnaires that are administered to the patients at baseline include both the primary and secondary outcome measures. In RedCAP, we have demographic data and items on co-morbidities as they are reported in the eligibility criteria.
Modifications of the protocol
For protocol amendments, we would first notify the sponsor and funder, then the PI will notify the center and a copy of the revised protocol will be added to the Investigator Site File. All deviations from the protocol will be fully documented using an adverse event report form. For amendments to the protocol, the protocol will be updated in the clinical trials registry. No substantial protocol amendments are anticipated, as no adverse events are expected. However, if an amendment becomes necessary—particularly one that affects patient safety, alters the scientific validity or scope of the study, or impacts ethical considerations—it must receive approval from the ethics committee of the canton Bern, Switzerland, before implementation.
Stopping rules
No stopping rules are applicable for this study.
Analysis and safety
The reporting of the study will follow the revised CONSORT statement [35].
Planned subgroup analyses
Subgroup analyses are planned for different age groups and for different surgical interventions (a: orthopedic metal removal; b: dressing/ change/ debridement/ wound flushing; and c: other minor surgical interventions).
Analysis plan and statistics tests
Descriptive analysis is used to compare baseline data to evaluate if the groups are comparable and that there were no problems/selection bias with the randomization procedure. In case there are patients that discontinued the intervention (VR) or there are missing values for the primary outcome, we will present an “intention-to-treat” and a “per-protocol” analysis. However, conclusions will be based on the “intention-to-treat” analysis. Pain scores are calculated according to the respective rules. Pain is measured by the VAS in children over 10 years and in children 6 to 9 years by the visual face scale. This ensures that it is the same outcome measure with the only difference in the age-adapted measuring instruments. If the primary and secondary outcome data do not follow the normal distribution, we compare the results between treatment groups (intervention (VR) vs. control group (standard pain therapy)) using the Mann–Whitney U test. In the event that the data follow the normal distribution, we will use Student’s t-test. Effect size d, the standardized differences between the two groups, will be calculated as described by Cohen [36]. For the main analysis, we will compare the results between treatment groups for the whole cohort. In an additional analysis, we will analyze each subgroup that is formed by the indication (a: orthopedic metal removal; b: dressing/change/debridement/wound flushing; and c: other minor surgical interventions) separately. This gives us the opportunity to evaluate whether the effect of the intervention is similar in these groups. No imputation techniques are used if secondary outcomes are missing. All p values will be reported one-tailed; no corrections will be made for multiple comparisons.
Interim analysis
An interim analysis is not planned.
Data monitoring committee
As the treatment group includes the use of commercially available VR games which has been used in previous studies, it is not expected to exert any harm to the patient. As such, only minimal monitoring is necessary. This includes a check that all informed consent forms have been dated and signed and a data validation check before data analysis. This monitoring will be performed by an external moderator. We will contact either an external study nurse or the CTU of the University of Bern regarding this issue.
As part of the study implementation, the project group management meets once a month to discuss the study implementation and any problems and questions that may arise. The trial steering group, established by the sponsor, monitors the progress of the study throughout its implementation with the PI and the data storage provider, as well as through annual reports to the ethics committee. A data monitoring committee was not implemented for the study, which was classified as low risk.
External validity
Given the nature of this study design, where the study is performed in a busy department consisting of several surgeons, the external validity is higher than it would be if the study was performed by a single pediatric surgeon. In addition, the high number of cases and the large geographical area from which patients are referred enhance the external validity. However, certain restrictions regarding the assessment of external validity still apply.

留言 (0)