
記住我
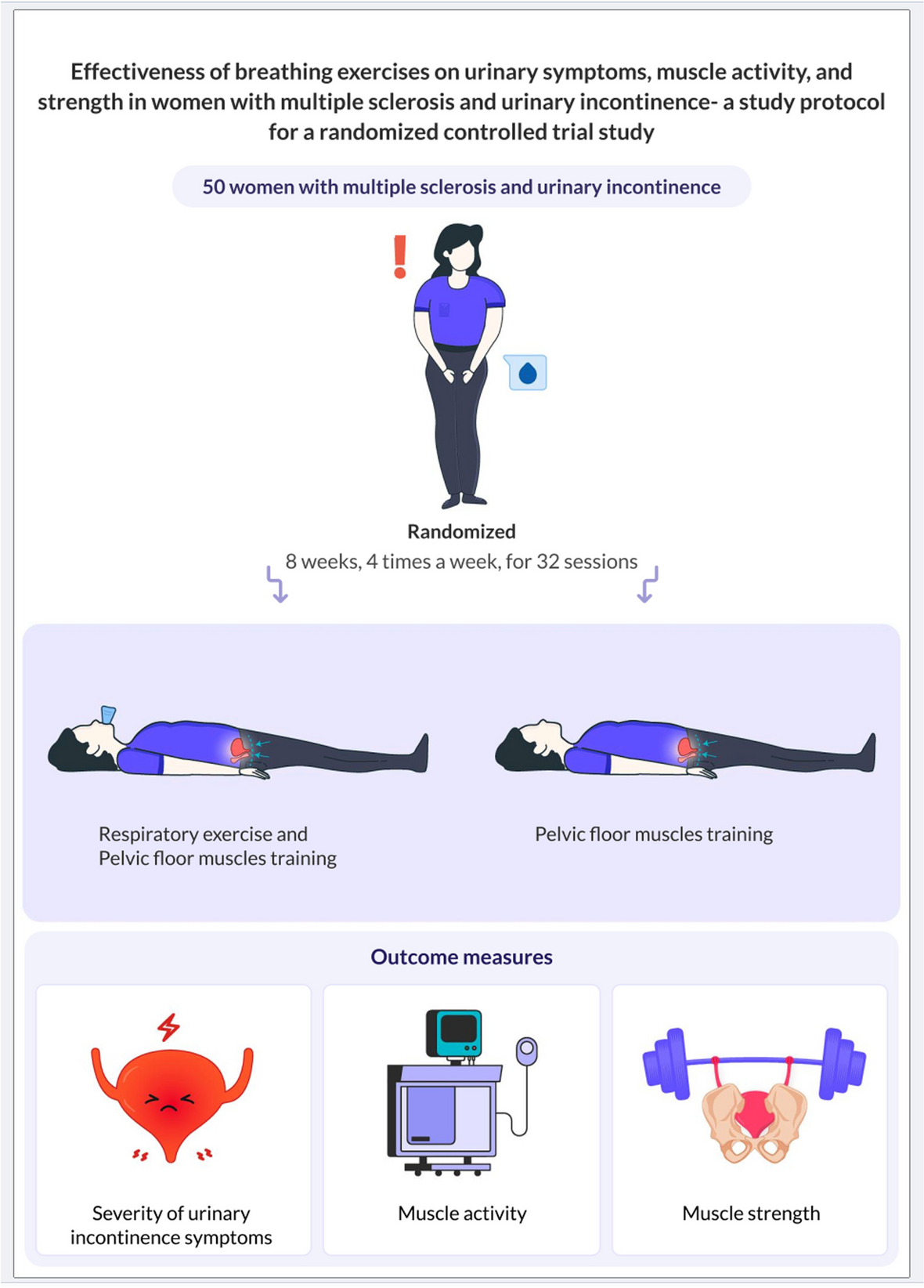
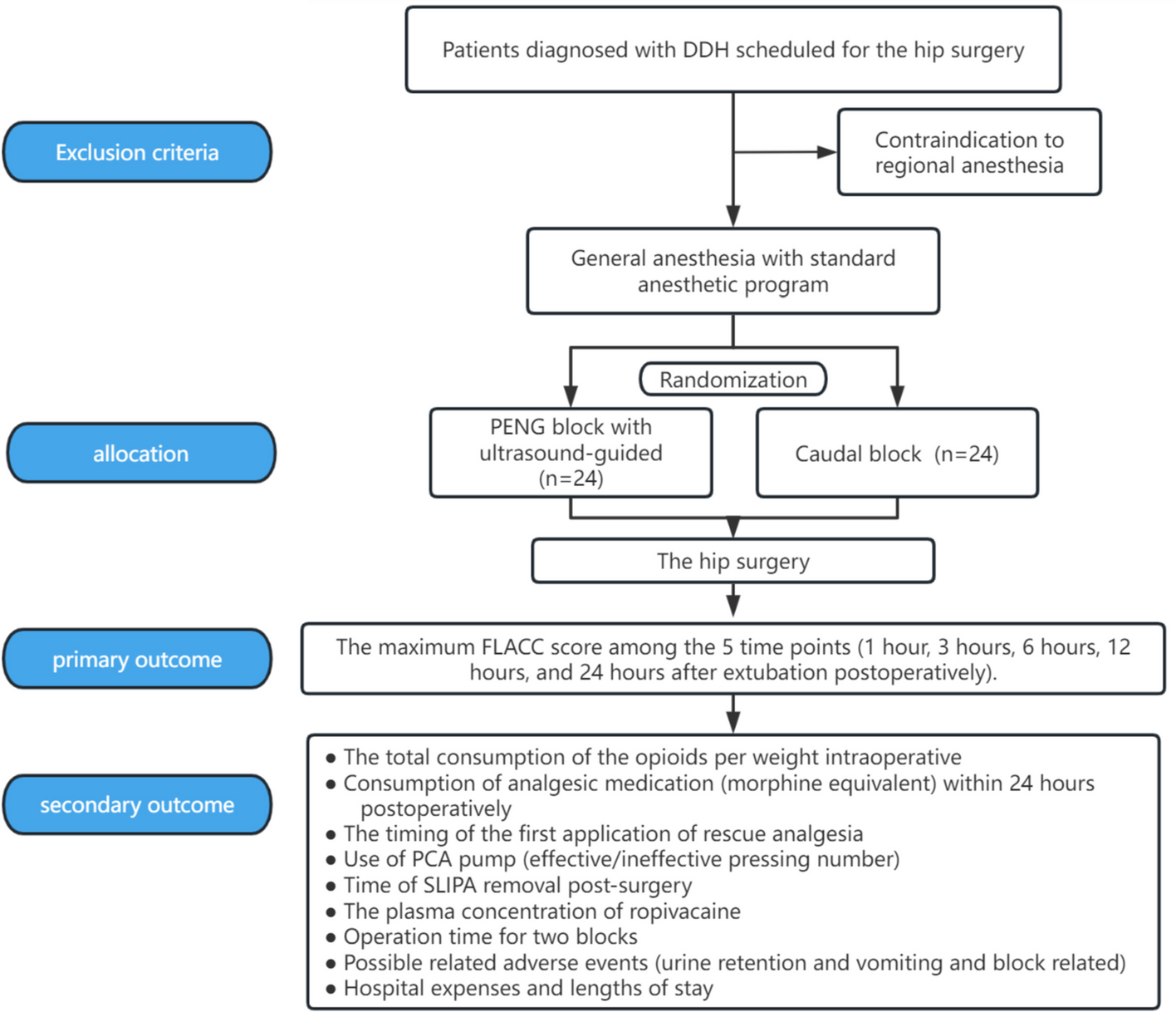
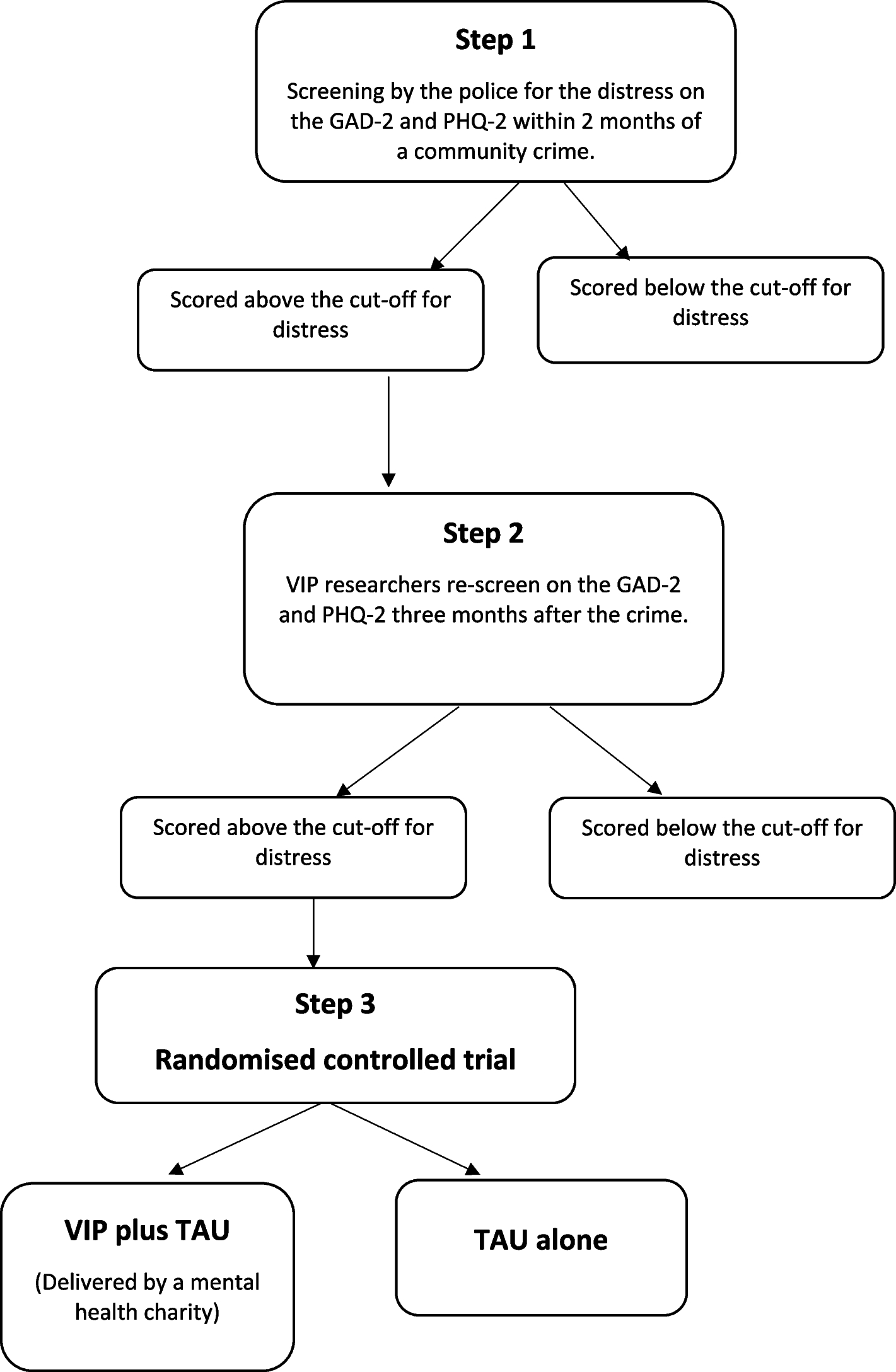
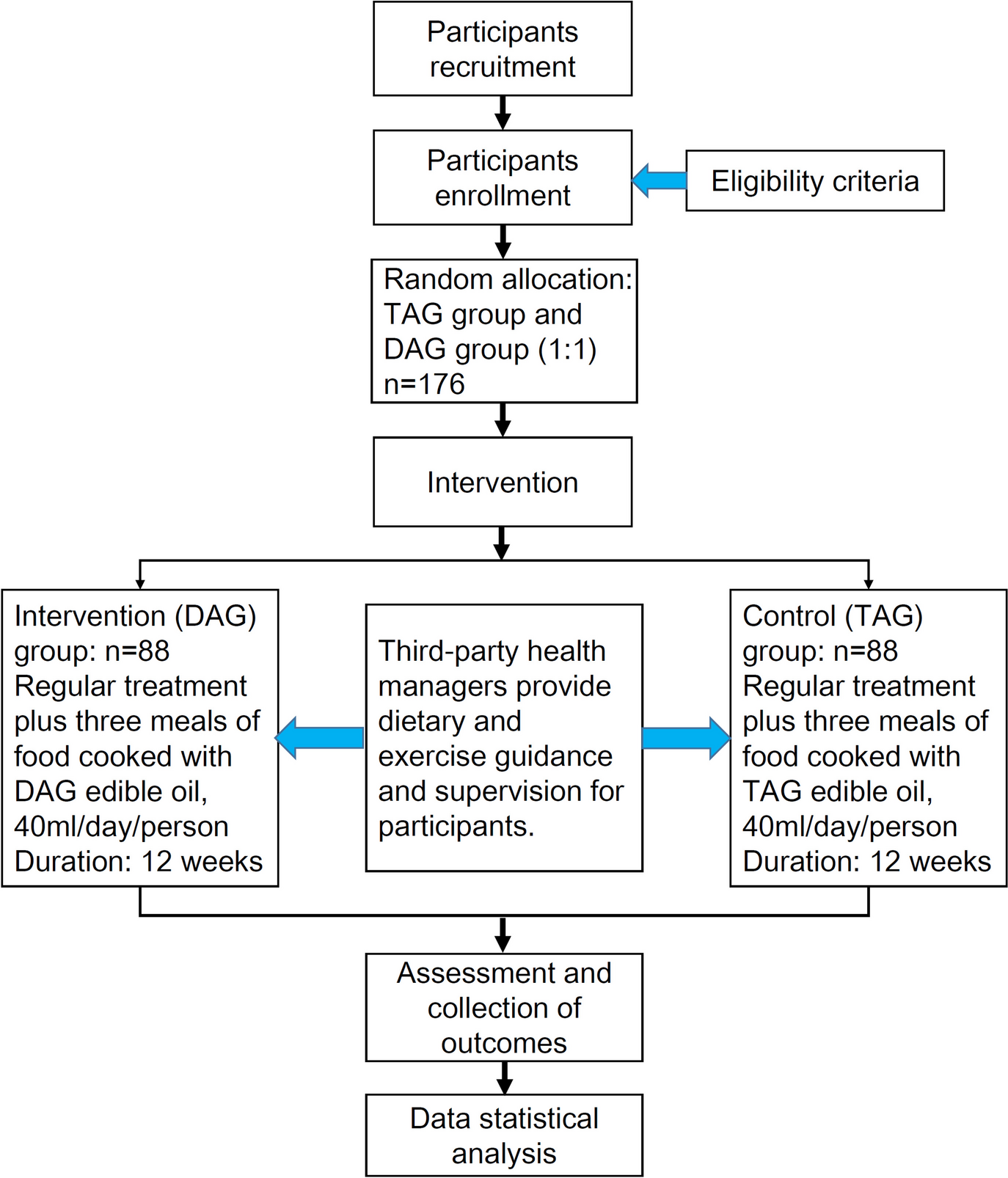
Spironolactone has been recommended as fourth-line treatment for patients with RH. In order to improve patient compliance and reduce experimental errors, the control group in this trial was standardized to use two tablets of OA + indapamide 2.5 mg + spironolactone 20 mg (A + C + D + spironolactone for short) once a day.
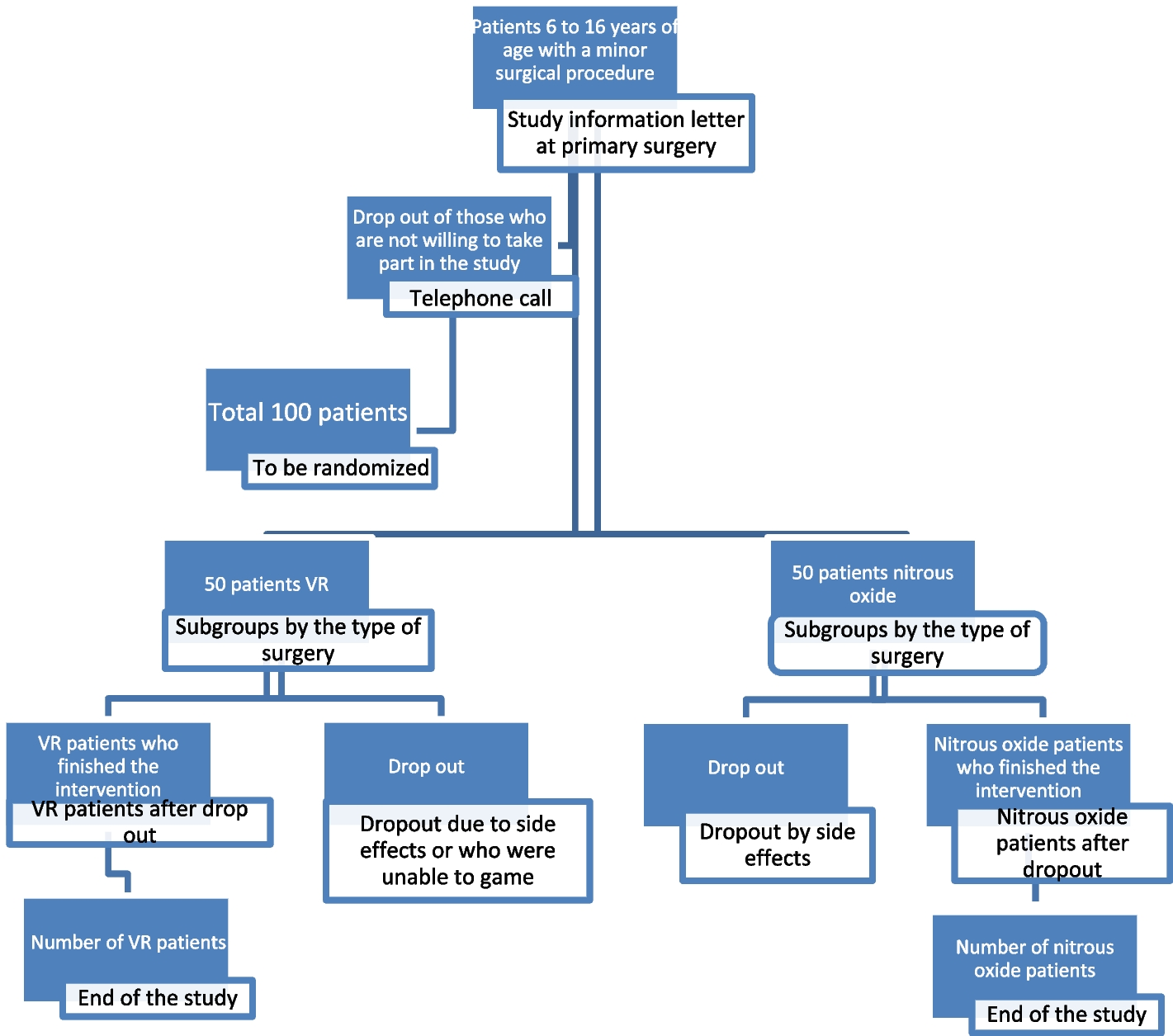
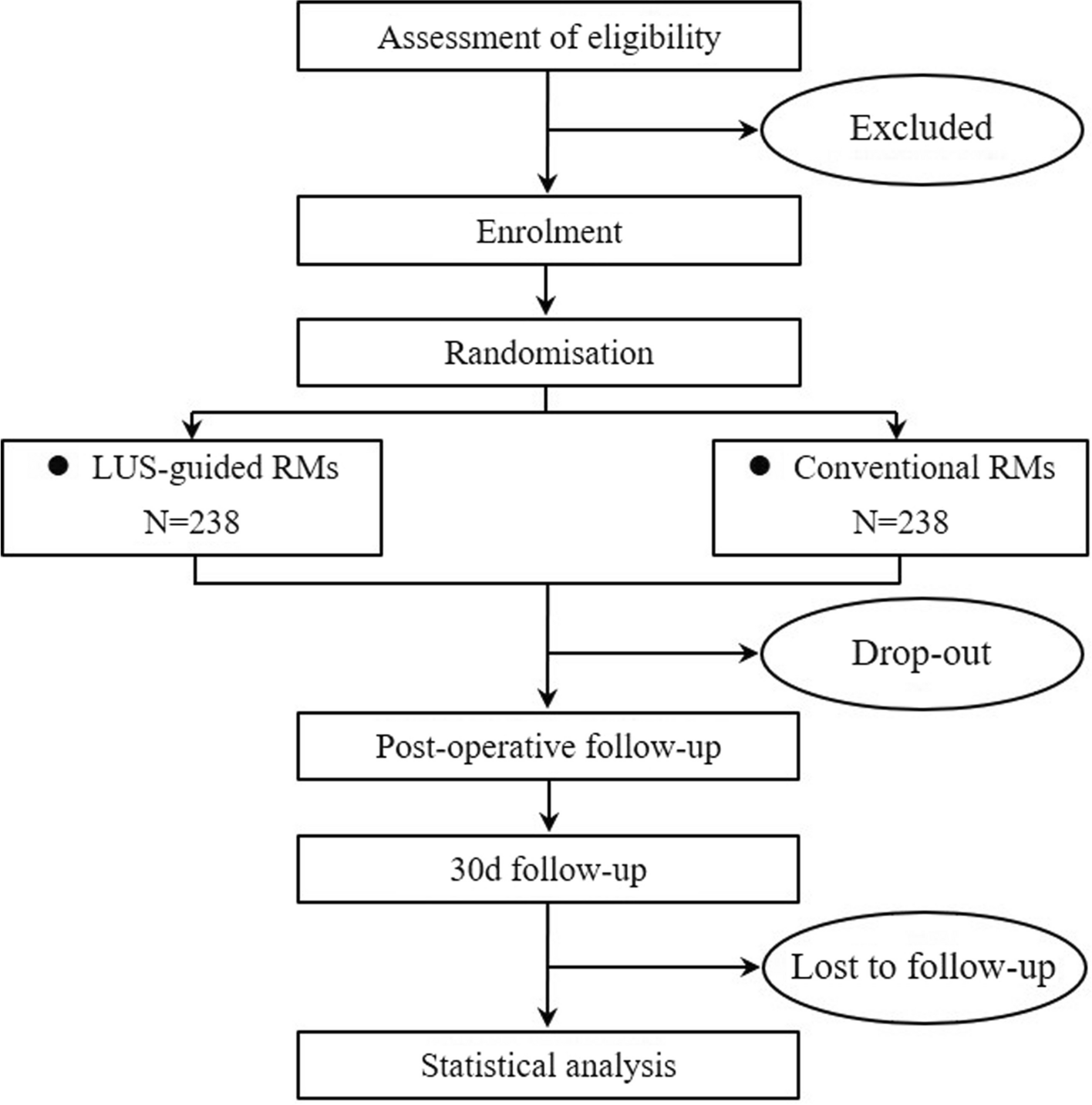
Intervention descriptionPatients with RH who met the above criteria were included in the study. After a screening visit, patients eligible for randomization proceeded to a 2-week import period with A + C (two tablets of OA daily). After the import period, the participants enrolled will be randomized into two crossover groups at a 1:1 ratio. One group will be given two tablets of OA + 1 tablet of compound reserpine and triamterene tablets (A + C + 0) once a day for 6 weeks, washed with A + C for 4 weeks, and then switched to the standard treatment regimen (A + C + D + spironolactone) for 6 weeks. The other group will be given the standard treatment regimen (A + C + D + spironolactone) for 6 weeks, washed with A + C for 4 weeks, and then switched to A + C + 0 for 6 weeks. We will closely monitor the changes in BP and associated adverse events in the two treatment groups during the two phases. Figure 1 illustrates the diagram of the study.
Fig. 1
The diagram of this trial
Determination of the wash-out period: according to the half-lives of the 4 drugs (30–50 h for OA, 14–24 h for indapamide, 9–16 h for spironolactone, and 45–128 h for compound reserpine and triamterene tablets), the wash-out period of this study was 4 weeks, on 3 to 5 half-lives.
In our study, A + C: two tablets of OA (olmesartan 40 mg, amlodipine 10 mg), 0: one tablet of compound reserpine and triamterene tablets, D: indapamide 2.5 mg, spironolactone 20 mg.
Criteria for discontinuing or modifying allocated interventionsAll patients could voluntarily participate in and withdraw from the study at any time without any reason or consequences. The trial will be discontinued if the patients have a serious adverse reaction.
Strategies to improve adherence to interventionsTo improve adherence, patients enrolled will be contacted weekly by phone or text messages to remind them to take medication. Research team members will conduct pill counts and help patients complete the eight-item Morisky Medication Adherence Scale (MMSE-8) to assess medication compliance at each face-to-face follow-up visit. Medication compliance = (Number of doses prescribed − Number of doses recovered)/Number of doses prescribed × 100%.
Relevant concomitant care permitted or prohibited during the trialThe use of any other antihypertensive medication or medication that affects blood pressure was prohibited during the trial.
Provisions for post-trial careAll participants will continue to be followed up in hypertension outpatient clinics. If participants suffer any harm from the trial, they will be treated according to the standard medical procedures.
Outcomes Primary outcomesThe primary objective will be to determine the reduction in average 24-h systolic blood pressure after 6 weeks of intervention between the compound reserpine and triamterene tablets treatment regimen and the standard treatment regimen in RH.
Secondary outcomes 1.Nighttime and daytime mean ambulatory blood pressure
2.Office blood pressure
3.Morning BP surge in ambulatory blood pressure monitoring (ABPM)
4.BP control rate
5.Anxiety/depression scale scores
Safety evaluationThe safety evaluation will include the incidence of adverse events (AEs, defined as any intervention-related unfavorable medical outcomes, including gastrointestinal reactions, hypotension, dizziness, headache, falls, syncope, insomnia, and acute gout attack), serious adverse events (SAEs, including hospitalization, prolonged hospitalization, disability, life-threatening, or death during the trial), and changes in biochemistry results (including electrolyte disturbance, hyperuricemia, and deterioration of kidney function).
The detailed definitions of safety outcomes are as follows:
(1)Gastrointestinal reactions, such as bloating, abdominal pain, nausea, vomiting, and belching;
(2)Hypotension, defined as SBP < 90 mmHg or DBP < 60 mmHg;
(3)Dizziness, defined as a false sense that you or your surroundings are spinning or moving;
(4)Headache, defined as a pain in the head or face;
(5)Falls, defined as sudden involuntary changes in body position;
(6)Syncope, defined as a transient loss of consciousness due to transient global cerebral hypoperfusion characterized by rapid onset, short duration, and spontaneous complete recovery;
(7)Insomnia, defined as the condition of being unable to sleep over a period of time;
(8)Acute gout attack, defined as a form of arthritis that causes sudden attacks of tenderness and pain in the joints;
(9)Electrolyte disturbance, including hyperkalemia (serum potassium > 5 mmol/L), hypokalemia (serum potassium < 3.5 mmol/L), hypercalcemia (serum calcium > 2.75 mmol/L), and hyponatremia (serum sodium < 125 mmol/L), etc.;
(10)Hyperuricemia, defined as serum uric acid > 420 μmol/L in men or postmenopausal women, or > 360 μmol/L in women;
(11)Deterioration of kidney function, defined as ≥ 50% reduction in eGFR in patients with CKD at baseline, or ≥ 30% reduction in eGFR to < 60 ml/ min/1.73 m2 in patients without CKD at baseline, or eGFR < 30 ml/min/1.73 m2.
Measurement of outcomes 24‑h BP measurementIn this study, ABPM readings were obtained from the nondominant arm using a TM2430 BP monitor (A&D Inc., Tokyo, Japan). Trained nurses will choose suitable size cuffs fitting the participant’s arm circumference to record 24-h BP. Before wearing the cuff, the researchers will explain the precautions to the participants, emphasizing the necessity to maintain their daily routines, avoid vigorous exercise, and remain still during each blood pressure measurement. During monitoring, BP will be measured every 20-min intervals during the day (from 6.00 a.m. to 10.00 p.m.) and every 30 min at night (from 10.00 p.m. to 6.00 a.m.). Reliable ambulatory blood pressure monitoring requires that the number of effective BP readings should reach more than 70% of the total number of monitoring times. Additionally, BP readings should be taken at least once an hour, with at least 20 readings during daytime and at least 7 readings during nighttime. Upon completion of each 24-h recording session, the data were uploaded from each site to the web-based Shuoyun system (https://www.heilcloud.com). Analysis and reporting were conducted in a standardized manner according to the current guidelines by ABPM technologists from West China Hospital.
Office BP measurementOffice BP will be measured via calibrated electronic sphygmomanometers (HBP-9020, Omron Corp., Kyoto, Japan). To obtain accurate BP data, all participants were requested to avoid smoking, drinking coffee, or exercising within 30 min. And they will empty their bladder and rest for 5 min in a chair with a backrest before the measurement. BP should be measured in both upper arms at the initial visit, and the side with the higher SBP reading will be the measured upper arm. The measurement of BP should be repeated 1 ~ 2 min apart, and the average of the two readings will be the final BP value. If the two readings of SBP or DBP differ by more than 5 mmHg, a third measurement should be taken and the average of the three readings will be used in the final analysis.
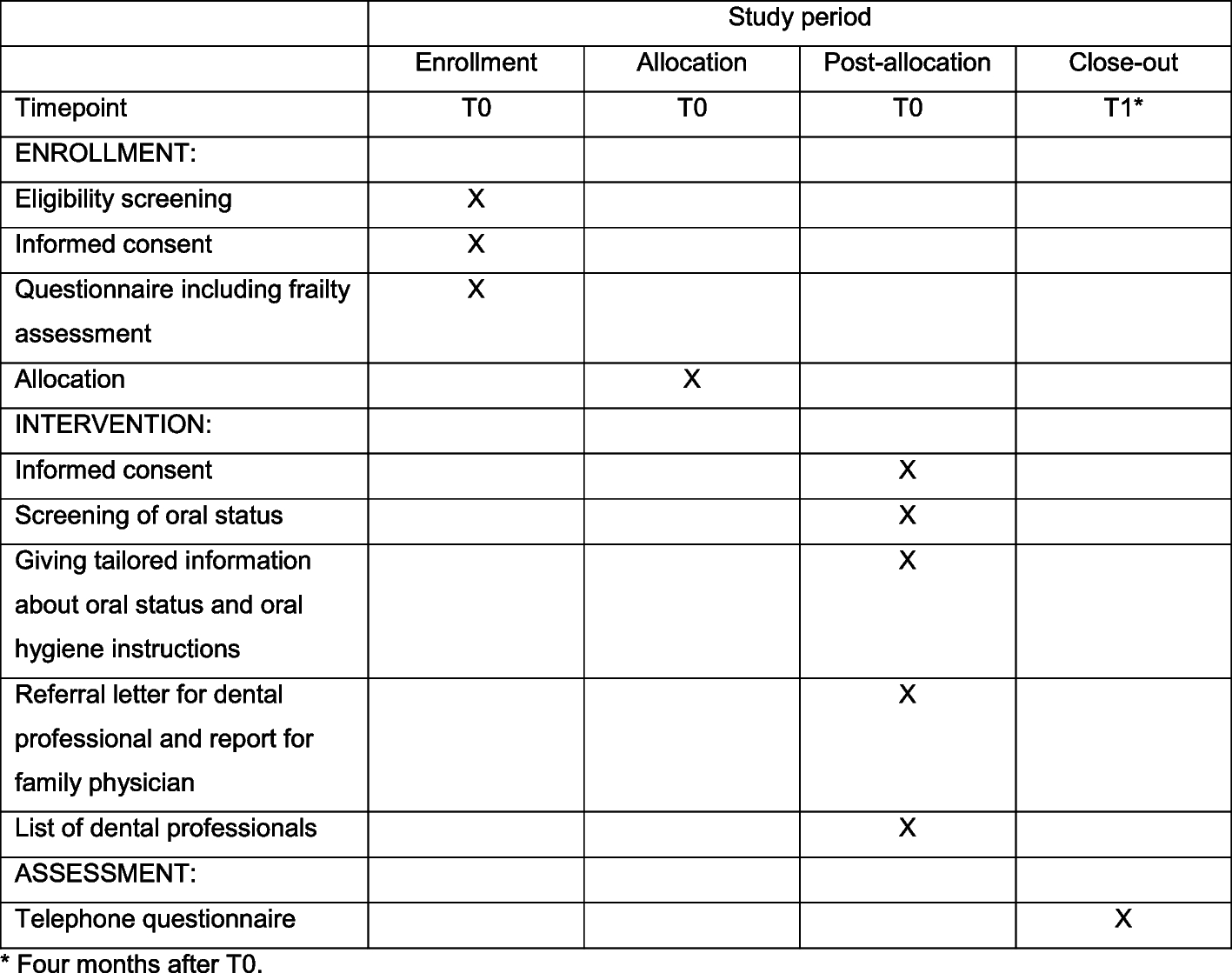
Participant timelineSee Table 1.
Table 1 Schedule of enrolment, interventions, and assessmentsSample sizeTo assess the noninferiority of the intervention group to the standard treatment regimen in terms of the primary efficacy outcome, the margin of noninferiority was set to 2.5 mmHg in the current study. Considering the differences between individuals and the allowable error of blood pressure monitoring, a blood pressure difference of 5 mmHg is common in clinical settings. To be conservative, after discussion with clinical experts, half of the 5 mmHg was adopted as the margin in the current study. Few studies have evaluated the antihypertensive efficacy of spironolactone versus compound reserpine and triamterene tablets in the same trial in patients with RH. Hence, we empirically estimated a difference of 5 mm Hg in systolic BP between treatment groups with a standard deviation SD of 15 mm Hg. The power is set at 80% (one-sided alpha level of 0.025) in the trial. We use the following formula, which is specially for sample size calculation of crossover noninferiority design to calculate the total number. The result is n = 16. We also calculate the sample size via PASS version 15.0, with the sample size of n = 17. Considering 15% loss to follow-up, a final sample size of 40 participants with 20 in each crossover group is requested in the trial.
$$n=\frac_+_\right)}^_}^}^}$$
n = sample size in each group
α = type 1 error = 0.025
β = 1-power = 0.2
\(_\) = the standard deviation of the difference in SBP reduction between the two treatment groups
\(\varepsilon\) = the difference between mean systolic blood pressure values of the two treatment groups
\(\Delta\) = noninferiority cutoff (\(\Delta\)<0)
RecruitmentPotential patients with RH will be identified from the hypertension outpatient clinics and inpatient department. We will invite them to participate by phone or face-to-face communication.
留言 (0)