
記住我

TAG is the main component of edible oil in daily life, including soybean oil, peanut oil, olive oil, rapeseed, and camellia oil, and is often used in comparative studies with DAG oil [23].
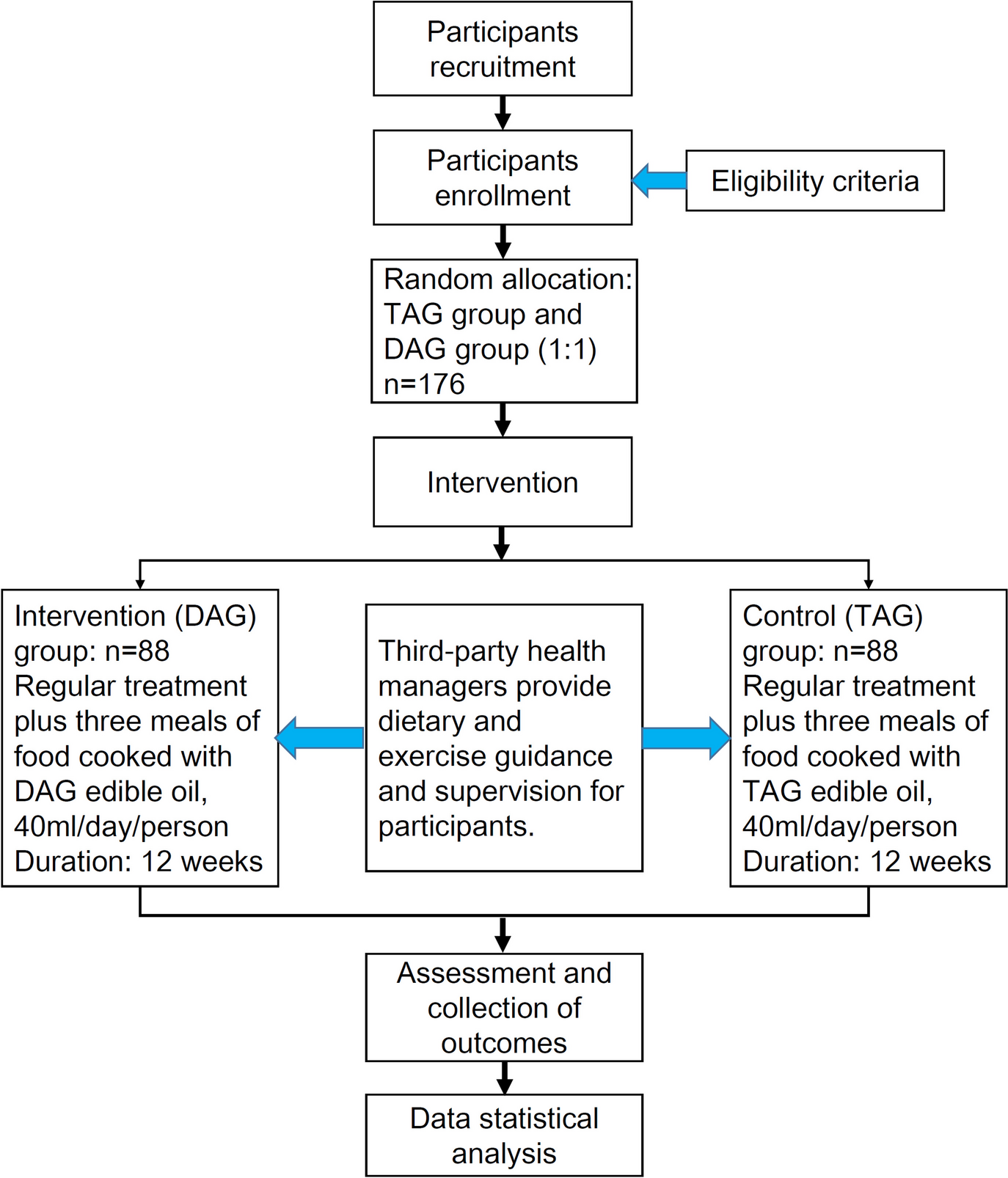
Intervention descriptionIntervention group: regular treatment plus three meals of food cooked with DAG edible oil, 40 ml/day/person. Control group: regular treatment plus three meals of food cooked with TAG edible oil, 40 ml/day/person. Regular treatment includes pharmacotherapy for hypertension. Therapy duration: continuous use for 12 weeks. During the trial, the test cooking oils are provided free of charge by the project team. Concurrent use of medications outside the scope of this trial for treating the condition is prohibited. All participants maintain a healthy lifestyle, including weight control, regular exercise, dietary adjustments to reduce caloric intake, smoking cessation, and limiting alcohol, high-purine, and high-fructose diets. Each participant will be provided with a measuring cup to ensure a daily oil intake of 40 ml DAG or TAG oil. Dietary guidelines will be offered to each participant to ensure balanced intake of energy and fats.
Dietary guidelines: refer to “Chinese Dietary Guidelines (2016)” [24]:
Diversify food intake with a focus on grains.
Balance diet and physical activity to maintain a healthy weight.
Increase consumption of vegetables, fruits, and dairy.
Consume fish, poultry, eggs, and lean meats in moderation.
Reduce salt, oil, sugar intake, and limit alcohol.
Criteria for discontinuing or modifying allocated interventionsWhen adverse events occur, the safety of participants should be put in the first place. According to the nature and severity of adverse events, different treatment methods such as termination of clinical trial, dose adjustment, symptomatic treatment, and observation can be adopted. In the event of withdrawal, investigators will communicate with the subject and complete the next scheduled visit content on the day of the participant’s decision to withdraw. If a participant refuses further visits, the last available information before withdrawal will be used for safety and efficacy evaluations. Investigators should record the reasons for withdrawal in detail and document them in the case report form (CRF).
Withdrawal at investigator’s discretionAn investigator may decide to withdraw a subject from the trial if it is deemed unsafe or inappropriate for the subject to continue. The following circumstances warrant withdrawal:
(1)Lack of symptom improvement, symptom exacerbation, or emergence of new severe symptoms (e.g., chest distress, palpitations).
(2)Allergic reactions or serious adverse events that, in the physician’s judgment, necessitate trial discontinuation.
(3)Poor subject compliance (product compliance < 80% or > 120%), or self-initiated changes to medication or use of prohibited drugs (considered withdrawal at the time of change).
(4)Post-randomization discovery of serious violations of inclusion or exclusion criteria.
(5)Participants or their guardians unwilling or unable to continue the trial for any reason, requesting withdrawal.
(6)Participants not explicitly requesting withdrawal but becoming lost to follow-up by not accepting product use and testing.
Participant-initiated withdrawalParticipants have the right to withdraw from the trial at any time, as stipulated in the informed consent form. Withdrawal is also considered to have occurred if a participant ceases to accept product use and testing, even without explicit request to withdraw. Reasons for withdrawal should be explored and documented, such as perceived lack of efficacy, intolerability of adverse effects, inability to continue clinical research, financial concerns, or unexplained loss to follow-up.
Strategies to improve adherence to interventionsDuring the intervention, all participants will be asked to record their daily oil consumption, diet and exercise, and to have their photos taken with a dietitian. Daily reminders will be sent to each participant via instant message to inform them. In addition, all participants will be given written and oral instructions for each meal plan. Also, researchers will meet with them monthly to encourage them to stick to the eating plan.
Relevant concomitant care permitted or prohibited during the trial Concurrent medications (1)Participants with chronic conditions may continue long-term medications.
(2)Continue regular treatment, including antihypertensive medications.
(3)Symptomatic treatments will be decided by researchers and recorded.
(4)All concurrent medications must be detailed in the CRF.
Prohibited concurrent treatments (1)Usage of unprescribed medications or methods for chronic metabolic syndrome and asymptomatic hyperuricemia will be prohibited.
(2)Usage of unmarketed drugs or other clinical trial medications will be also forbidden.
Provisions for post-trial careUnblinding will be performed at the end of the 12-week clinical study evaluation or when participants experience trial-related adverse events, including clinically significant laboratory abnormalities. After discussion by the research team, the participant will receive continued medical care at the sub-center where the respective clinical study is conducted.
Outcomes Primary and secondary outcomesThe primary outcomes will be the levels of serum uric acid during the intervention 4 weeks ± 3 days, 8 weeks ± 3 days, and 12 weeks ± 3 days. The secondary outcomes will be the levels of fasting blood glucose, 2 h postprandial blood glucose, total cholesterol, triglyceride, high-density lipoprotein cholesterol, low-density lipoprotein cholesterol, blood pressure, weight, body mass index, waist circumference, and average intima-media thickness of common carotid artery during the intervention 4 weeks ± 3 days, 8 weeks ± 3 days, and 12 weeks ± 3 days; fasting insulin, fasting C-peptide, 2 h postprandial C-peptide, glycosylated hemoglobin, and HOMA-IR during the intervention 12 weeks ± 3 days.
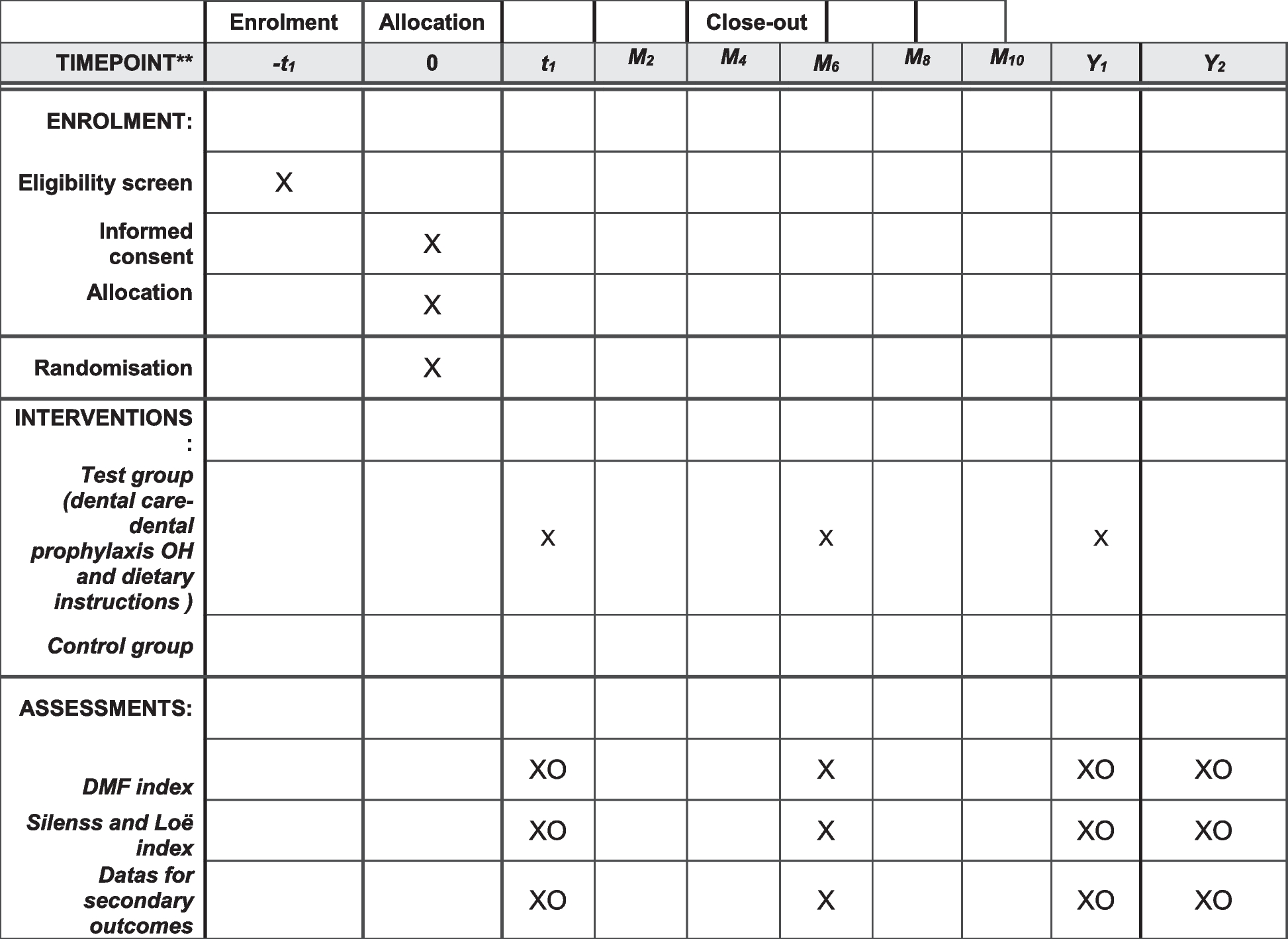
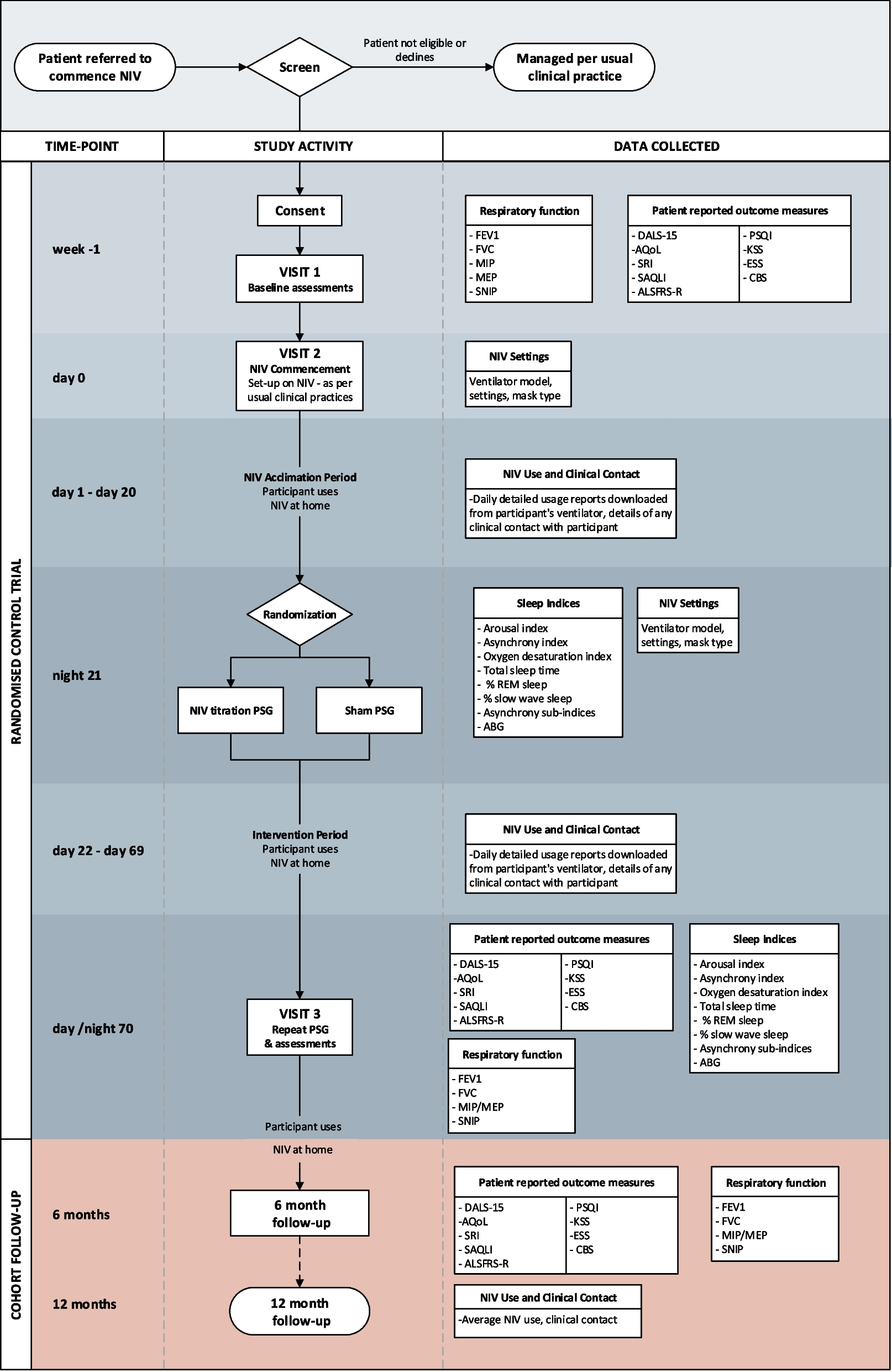
Participant timelineA time schedule of enrolment, interventions, and assessments is presented in Fig. 2.
Fig. 2
Schedule of enrolment, interventions, and assessments. at1, week 4 ± 3 days; t2, week 8 ± 3 days; t3, week 12 ± 3 days. bTC, total cholesterol; TG, triglyceride; HDL-C, high-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol
Sample sizeIn accordance with statistical principles, we utilized the PASS 2021 software’s Tests for Two Means in a Repeated Measures Design to calculate the sample size. Parameters were set based on preliminary trial results and expert consultation as follows: statistical power (1 − β) = 0.8, significance level (α) = 0.05, the ratio of sample sizes between the two groups = 1:1, expected difference in uric acid means between groups = 55 µmol/L, which is based on our previous small sample trial of diacylglycerol edible oil intervention in patients with chronic metabolic syndrome, a single arm trial, and the difference in mean before and after analysis after 2 months of intervention (not published in any journal), number of time points = 3, type of covariance = compound symmetry, standard deviation of a single observed sample = 137.99 µmol/L (range: 6.18 ~ 137.99), correlation coefficient between different time points = 0.55 (range: 0.23 ~ 0.55). The calculation determined that 70 cases per group are required, with an additional 20% to account for loss to follow-up, resulting in a final sample size of 88 cases per group. Consequently, the total sample size for this trial is 176 cases, with an equal ratio of intervention to control groups, i.e., 88 cases in each group.
RecruitmentWe will collect 176 participants from The Eighth Affiliated Hospital of Sun Yat-sen University, Shenzhen Hospital of Southern Medical University, The Third Affiliated Hospital of Guangzhou Medical University, and Shenzhen Hospital of Guangzhou University of Chinese Medicine. Recruitment posters containing inclusion and exclusion criteria will be posted in clinics and on the social networking platform. Interested participants will be invited to the hospitals for a physical examination and baseline assessment and will be informed that their participation is voluntary and that choosing not to participate will not affect their care.
留言 (0)