The current study was based on our previous findings in heart tissue from patients with advanced coronary artery disease and aortic valve disease, showing that diseased myocardium displayed increased expression of TLR1, TLR3 and TLR7, increased expression of downstream signaling mediators of these receptors, as well as markedly decreased expression of γ-protocadherins compared to control tissue [9, 10]. Thus, we were interested in investigating possible direct and specific effects of TLR1-, TLR3- and TLR7-activation on NF-κB signaling, proinflammatory cytokine production as well as γ-protocadherin expression.
Toll-like receptors (TLRs) are broadly expressed across various mammalian cell types, including dendritic cells (DCs), macrophages, neutrophils, monocytes, lymphocytes, fibroblasts, epithelial cells, endothelial cells (ECs), and nerve cells [13]. Notably, each cell type expresses specific TLR subsets that play distinct roles in recognizing pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs) and mediating the immune response [14].
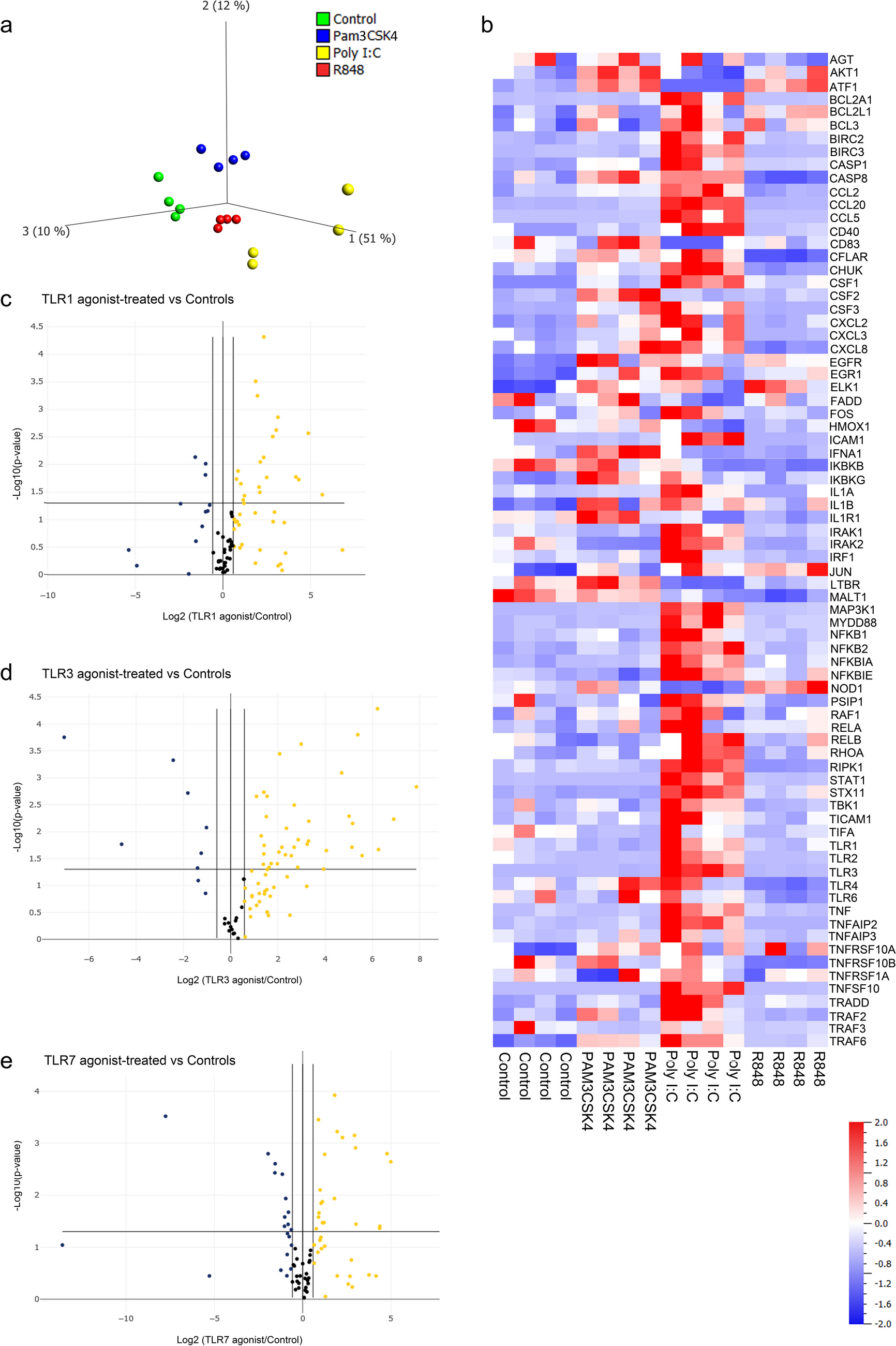
Cardiac tissue consists of many different cell types and fibroblasts constitute a significant proportion of the total cell population in the heart. Estimates suggest that fibroblasts make up approximately 15–20% of the total cell number in the adult mammalian heart [4, 15,16,17]. This proportion, however, can vary depending on the species, age, and physiological or pathological conditions of the heart. Cardiac fibroblasts are vital to heart function, playing key roles in maintaining ECM homeostasis, providing a structural scaffold for cardiomyocytes, distributing mechanical forces throughout the cardiac tissue, and mediating electrical conduction [4, 18, 19]. While cardiac fibroblasts are known to express Toll-like receptors (TLRs) and respond robustly to proinflammatory stimuli [1], cardiomyocytes exhibit a comparatively muted response to such stimuli [20, 21]. In murine models, cardiomyocytes have been shown to be selectively responsive to activation by TLR2, TLR4, and TLR5, whereas activation by TLR3 and TLR7 did not result in the downstream expression of cytokines and chemokines (TLR1 was not investigated) [22]. Therefore, it is likely that fibroblasts significantly contribute to the increased expression of TLR1, TLR3, and TLR7 and their downstream signaling pathways we previously observed in the myocardium of patients with cardiac disease [9]. The data from the current study demonstrate that cardiac fibroblasts indeed respond to activation by TLR1, TLR3, and TLR7, leading to downstream NF-κB signaling. Interestingly, principal component analysis (PCA) revealed that these receptors activate distinct genetic features within this pathway. Specifically, we found that TLR3 regulate NF-κB mediators as well as γ-protocadherins at the gene level more potently and uniquely in cardiac fibroblasts compared to TLR1 and TLR7. Additionally, TLR1 and TLR3 activation strongly induced the secretion of proinflammatory markers by cardiac fibroblasts, reaching levels similar to those observed with LPS stimulation. In contrast, TLR7 activation resulted in a more selective induction of cytokine secretion. Notably, IL-10 was secreted in substantial amounts in response to all agonists, even though its gene expression was undetectable. This discrepancy might be explained by post-transcriptional regulation through microRNAs and RNA-binding proteins, which degrade or inhibit mRNA, reducing its levels without impacting existing IL-10 protein. Additionally, IL-10 may have strong protein stability and a long half-life, or might be efficiently translated, resulting in high protein levels even from minimal mRNA presence.
TLR1 forms heterodimers with TLR2 and is activated by triacylated bacterial lipoproteins, including the synthetic triacylated lipopeptide Pam3CSK4, which mimics the acylated amino terminus of proinflammatory bacterial lipopeptides [23]. Although TLR1 has not been directly linked to cardiovascular disease (CVD), one study demonstrated that TLR2 activates cardiac remodeling in a TLR1-dependent manner in a heart failure mouse model [24].
Activation of TLR7 has been shown to be essential for miR146a-5p-induced myocardial inflammation and cardiomyocyte dysfunction in mice [25], and both human and mouse cardiac tissue exhibit upregulated TLR7 gene expression following myocardial infarction (MI) [26]. Overactivation of TLR7 signaling induces severe hemorrhagic myocarditis in mice [27], while TLR7 deficient mice display reduced adverse cardiac remodeling and improved cardiac function [26]. Additionally, TLR7 knockout mice are protected against myocarditis following the induction of autoimmune myocarditis [28].
Specific ligands for TLR3 include viral double-stranded RNA (dsRNA) and its synthetic analogue polyriboinosinic (Poly I:C) [29]. Increasing evidence indicates that TLR3 plays a crucial role in the initiation and progression of cardiovascular diseases (CVDs). TLR3 has been implicated in mediating sterile pathologies in CVDs associated with immune responses, including pulmonary artery hypertension (PAH), atherosclerosis, myocardial infarction (MI), ischemia/reperfusion (I/R), and heart failure [30,31,32]. Mouse studies have demonstrated the role of TLR3 in cardiovascular disease and tissue repair. For example, TLR3 knockout mice or pharmacological inhibition of TLR3 in vivo prevented the development of aortic valve stenosis [33]. Conversely, TLR3 activation has been shown to be essential for repair and regeneration in damaged neonatal mouse hearts, promoting glycolysis and cardiomyocyte proliferation through downstream YAP1 signaling [34].
TLR3 is unique among the Toll-like receptors (TLRs), mediating its downstream signals independently of the adaptor protein MyD88, which is a central signaling mediator for all other TLRs. The potent molecular effects of TLR3 on NF-κB mediators and γ-protocadherins in cardiac fibroblasts may rely on this distinct MyD88-independent pathway. This pathway involves the recruitment of the adapter protein Toll-IL-1 receptor (TIR) domain-containing adapter (TRIF, also known as TICAM-1) and downstream association with receptor-interacting protein 1 (RIP1), TRAF3, and/or PI3K [35,36,37,38]. Indeed, angiotensin II-induced hypertension and myocardial hypertrophy in mice have been shown to depend on, and be regulated by, TLR3-TRIF signaling [39]. Furthermore, Tang et al. demonstrated that TLR3 mediates myocardial hypertrophy through increased binding to S-nitrosylated muscle LIM protein and RIP3 (receptor-interacting protein kinase 3) and the activation of the NLRP3 (NOD-like receptor pyrin domain containing 3) inflammasome [40]. However, TLR3 has been shown to signal through MyD88 in specific cases, such as during neuronal development, where TLR3 negatively regulates the expression of Disrupted in schizophrenia 1 (Disc1) in cultured neurons and mouse brain studies [41]. In our study, activation of TLR3 by Poly I:C led to a strong increase in MyD88 expression, suggesting that TLR3 may signal through MyD88 in cardiac fibroblasts, or alternatively, that MyD88 functions as a negative feedback mechanism, acting as an inhibitor of TLR3/TRIF activation, similar to findings observed in mouse corneal epithelium by Johnson et al. [42].
We have previously observed that patients with advanced coronary artery disease or aortic valve disease exhibited reduced expression of several γ-protocadherin (γ-Pcdhs) genes in myocardial tissue compared to control samples [10]. We hypothesized that this reduction was linked to the upregulated expression of TLR1, TLR3, and/or TLR7 in the same tissue samples. γ-Pcdhs are a subfamily of the cadherin superfamily, well-known for their roles in neural development and cell–cell adhesion. Whereas other cadherins have been implicated in inflammatory processes, Pcdhs are still vastly unexplored in this context. However, some studies suggest an involvement of Pcdhs in inflammatory responses, for example in PcdhgC3 KO brain microvascular endothelial cells, which showed an increased induction in IL-6 following oxygen/glucose deprivation or TNFα treatment compared to wildtype cells [43]. Furthermore, PCDHA4 levels were reduced in extracellular vesicles released from thrombin-activated platelets, while increased in platelet membranes [44]. Human monocyte-derived macrophages transfected with the S protein of SARS-CoV-2 resulted in increased IL-1β, IL-6, and TNFα protein levels, accompanied by a decreased gene expression of PCDHs [45]. Additionally, the possible role of Pcdhs in cardiac morphogenesis and cellular signaling during heart development further underscores a potential role in cardiomyopathy associated with inflammation [46].
Our data in the current study suggest that PCDHGs are more significantly regulated at the gene level by TLR3 stimulation in cardiac fibroblasts compared to TLR1 or TLR7 stimulation. This regulation occurs independently of miR-133A/B, miR-448, and miR518C that we previously found associated with decreased PCDHG expression in diseased myocardium. The absence of these miRNAs in fibroblasts suggests that their influence on PCDHGs pertains to other cardiac cell types, such as cardiomyocytes or endothelial cells. Thus, PCDHGs in fibroblasts appear to primarily be regulated by TLRs through mechanisms not involving miR-133A/B, miR-448, or miR518C. It is important to note that other mechanisms may also regulate PCDHG expression, e.g., hypoxia, oxidative stress, mechanical stress, extracellular matrix changes, and growth factors like TGF-β, which are particularly relevant in the context of cardiac remodeling and fibrosis. These aspects warrant further investigation in future studies.
In summary, our findings indicate that TLR1, TLR3, and TLR7 activate distinct genetic features of the NF-κB signaling pathway in cardiac fibroblasts, with TLR3 emerging as a more potent activator compared to TLR1 and TLR7, particularly in suppressing PCDHG gene expression. Our data support a role for TLR3 in cardiac fibroblasts in contributing to the enhanced inflammatory state and reduced PCDHG expression previously observed in the diseased myocardium of patients with advanced coronary artery disease and aortic valve disease. Thus, TLR3 represents a potential therapeutic target for modulating immune responses associated with CVD.



留言 (0)