2.1 Materials
Ascorbate 2-phosphate, β-glycerophosphate, paraformaldehyde (PFA), trypsin–EDTA, 2-amino-2-methyl-1-propanol (AMP), o-cresolphthalein complexon, 8-Hydroxyquinoline, Bovine serum albumin (BSA), Safranin O, Tween20, Kolliphor®P188 were obtained from Sigma (Taufkirchen, BY, Germany). L-Prolin, sodium-pyruvate, acetic acid, hydrochloric acid (HCl) and crystal violet were received from Roth (Karlsruhe, BW, Germany). Protease inhibitor cocktail, DAPI and insulin were bought from Roche (Mannheim, BW, Germany). Resazurin was purchased from Santa Cruz Biotechnology (Heidelberg, BW, Germany). Fetal bovine serum (FBS) was from Capricorn Scientific (Ebsdorfergrund, HE, Germany). ITS-G premix, high / low glucose medium (4.5 g/L) and Dulbecco’s Modified Eagle’s medium (DMEM/F-12) were obtained from Life Technologies (Darmstadt, HE, Germany). ∆9-tetrahydrocannabinol (Dronabinol) was bought from LGC (Wesel, NW, Germany). Collagenase (type I) was purchased from Worthington Biochemical Corp. (Lakewood, NJ, USA). Transforming growth factor-β3 (TGF-β3) was obtained from PeproTech (Hamburg, HH, Germany). 2-propanol and Oil Red O was bought from Merck (Darmstadt, HE, Germany).
2.2 Cell culture
Fourteen healthy female Caucasian patients undergoing elective abdominoplasty at the Department of Plastic Surgery, Hand Surgery–Burn Center at the University Hospital RWTH Aachen were recruited for the study. Informed consent was obtained by all patients. The study protocol was approved by the regional ethics committee (ethics committee of the RWTH Aachen University Faculty of Medicine, Aachen, Germany; EK163/07), and the reported investigations have been carried out following principles endorsed by the Declaration of Helsinki.
Adipose tissue samples used for isolation of ASCs were processed as described earlier [15]. Adipose tissue was minced and digested in collagenase solution (0.2% collagenase I in PBS) for 45 min at 37 °C. Digested tissue was filtered through a 250 nm nylon mesh (Neolab, Heidelberg, BW, Germany). Oil and cell debris as well as the mature adipocyte fraction were separated from the stromal vascular fraction (SVF) by centrifugation at 400 xg for 10 min. Afterwards, the SVF was resuspended in proliferation medium (DMEM, 10% FBS), seeded on culture plates and cultured at 37 °C, 5% CO2. From these primary cultures, experiments were conducted with cells from passages 1–4 seeded at a density of 3 × 104 cells per cm2.
2.3 Pharmacological stimulation
Because THC is characterized by high lipophilicity, it has to be diluted in an appropriate vehicle for application in aqueous solutions. Earlier studies investigating the effects of THC used non-aqueous solvents, emulsifiers or surfactant molecules, or mixtures of these agents as a vehicle. Therein, the surfactant molecule Kolliphor®EL (formally known as Cremophor EL) has been regularly applied as vehicle for better aqueous solubility and stability of THC [16]. However, we found that EL is not an inert vehicle but exerts a range of biological effects on ASCs, making it unsuitable for the planed experiments [17]. Therefore, we decided to use another surfactant, Kolliphor®P188 (also known as Poloxamer P188) does not affect ASC biology and has already been used as vehicle for THC solutions [18].
For a 1 mM solution, THC was dissolved from a stock solution in methanol into a vehicle solution consisting of saline with 7.5% methanol and 5% P188. Saline containing the respective concentration of methanol and P188 served as vehicle control (Veh). To ensure complete adherence of the cells, THC was freshly added to the media two days after seeding and during each media exchange.
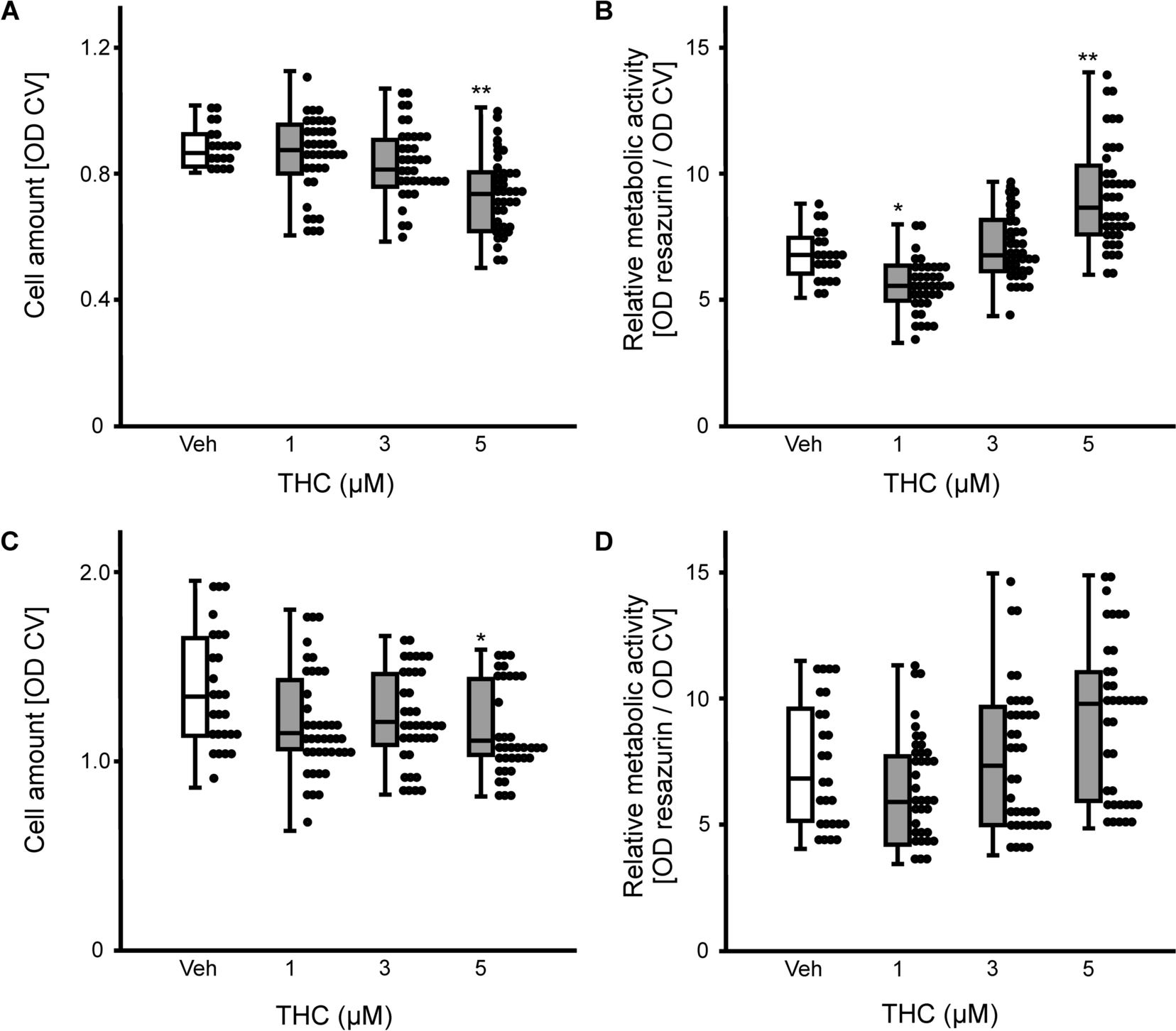
2.4 Cell viability
At the indicated time points, the cell viability was assessed by determining cell number using crystal violet (CV) staining and via measuring resazurin conversion by mitochondrial enzymes, which serves as an indicator of cellular metabolic activity. Each procedure followed the same regime as described earlier [13]. Cells were incubated with resazurin solution (final concentration 0.01 mg/mL in proliferation medium) for 45 min. Afterwards, fluorescence was measured at 590 nm (excitation = 544 nm) using a microplate reader (BMG Labtech, Ortenberg, HE, Germany). For CV staining, the cells were washed with PBS, fixed in 2-propanol followed by washing with 0.05% Tween20 in PBS. After staining the cells with 0.1% CV in aqua bidest, the adsorbed dye was washed out in 33% acetic acid. Absorbance was quantified at 595 nm.
2.5 Cell differentiation
Adipogenic differentiation was induced by high glucose medium supplemented with 10% FBS and 10 µM insulin. Osteogenic differentiation medium was low glucose medium with 10% FBS, 0.25 µM dexamethasone, 10 mM β-glycerophosphate, 200 µM ascorbate-2-phosphate. Chondrogenic differentiation medium contained DMEM supplemented with 5 ng/mL TGF-β3, 1 μM dexamethasone, 50 µg/mL ascorbate-2-phosphate, 40 µg/mL proline, 90 µg/mL pyruvate, and 1 mL ITS 1 Premix (1 mg/mL insulin, 0.55 mg/mL transferrin, 0.67 μg/mL sodium selenite).The medium was replaced every 2 to 3 days. Evaluation of trilinear differentiation was performed as described earlier [13]: Adipogenic differentiation was assessed by Oil Red O staining. ASCs were PFA fixed and stained with Oil Red O solution (0.3% in isopropanol:aquabidest, 3:2). Adsorbed dye was washed out with isopropanol and absorbance was measured at 540 nm. For evaluation of osteogenesis, PFA-fixed cells were incubated in cresolphthalein-buffer (50 mg o-cresolphthalein complexon and 500 mg 8-Hydroxyquinoline dissolved in 30 mL of 37% HCl, and diluted in 470 mL aquabidest). After adding AMP buffer (76 mL AMP in 440 mL aquabidest, pH = 10.7 with HCl), the extinction of the supernatant was quantified at 580 nm. Chondrogenic differentiation was measured by Safranin O staining. Differentiated cells were stained with 0.1% Safranin O aquabidest. The adsorbed dye was washed out with isopropanol and quantified at 540 nm.
2.6 Animals
Mice (C57BL/6 J; n = 18, female) were purchased from Janvier Labs (Tancon, France) and kept in the facilities of the Institute of Laboratory Animal Science at the University Hospital, RWTH Aachen. The mice were housed in groups of 4–5 animals in plastic cages with sawdust bedding and had access to food and water ad libitum. The animal housing rooms were kept at 21–24 °C and 40–60% relative humidity with a 12-h light/dark cycle. The animals were randomly divided into two groups and treated with either THC or vehicle solution, respectively, over a time course of 21 days. After daily determination of the bodyweight (BW), the animals received an injection (200 µL, s.c.) of either THC (3 mg/kg BW), or vehicle solution [16]. Mice were sacrificed for experimental investigation by cervical dislocation in deep anesthesia (5% isoflurane). The skin was disinfected with 70% ethanol, and adipose tissue was obtained from fully excised perigonadal and perirenal fat deposits for determination of growth factor contents as well as for histological analysis [12]. The animal experiments were performed according to EU Directive 2010/63/EU for animal experiments, they followed the guidelines of the animal welfare laws and were approved by the Animal Care and Use Committee of the state of North Rhine-Westphalia, Germany (AZ-81–02.04.2021.A352).
2.7 Histology
Adipose tissue of mice was cut into samples of ~ 2–3 mm side length, fixed in 4% PFA for 24 h and then embedded in paraffin. Slices of 2 µm thickness were prepared on a microtome (Hyrax M40, Zeiss, Oberkochen, BW, Germany), mounted on microscope glass slides and dried overnight in an incubator (37 °C). After dehydration in an ascending alcohol series and xylene, the slides were cover slipped with ROTI®Histokitt (Carl Roth, Karlsruhe, BW, Germany). In each slide, 3–5 randomly chosen regions of interest (ROI = 290 × 228 µm2) were photographed on an EVOS FL auto imaging system (Thermo Fisher Scientific, Waltham, MA, USA). The volume of lipid vacuoles was determined as indicator of fat cell size using the free software ImageJ (Wayne Rasband, Institutes of Health, Bethesda, Rockville, MD, USA).
2.8 Cytokine determination by enzyme-linked immunosorbent assay (ELISA)
After seven days of stimulus exposure (Veh or THC), the cell supernatant of each well from the viability experiments was collected to determine the concentrations of the hepatocyte growth factor (HGF), the transforming growth factor (TGF)‐β1 and the vascular endothelial growth factor (VEGF), by enzyme-linked immunosorbent assay ELISA Duo‐Sets (R&D Systems, Minneapolis, MN, USA) following the manufacturer's instructions. Extinction was measured in duplicates per well. Cytokine concentrations were expressed as the amount of each factor (in ng) per number of cells (OD crystal violet).
For the evaluation of cytokine content in adipose tissue in vivo, the concentrations of HGF, TGF-β1 and VEGF were determined. The adipose tissue was homogenized in 2 mL lysis buffer (pH = 7.5, 10 mM HEPES, 0.5% Triton X-100, protease inhibitor) on ice using an Ultra-Turrax tissue homogenizer (IKA Works, Inc, Wilmington, NC, USA). The homogenized suspension was first centrifuged at 1,000 xg for 10 min at 4 °C, followed by second centrifugation at 14,000 xg at 4 °C for 40 min. The oily phase and cell pellet (consisting of non-homogenized cells and nuclei) were discarded. The supernatant was divided into aliquots and taken for cytokine determination as described above. Cytokine concentrations were expressed as the amount of each factor per mg of soluble extracted protein. Whole protein concentrations of the probes were assessed by the DC Protein Assay (BioRad, Hercules, CA, USA) as specified by the manufacturer.
2.9 Statistical analysis
Data of all experiments were grouped to evaluate the results for each type of experiment and were tested for normal distribution using the Kolmogorov–Smirnov test. Normally distributed data were presented as mean values (+ SEM) and were statistically analyzed using Student’s t-test or by analysis of variance (one-way or two-way ANOVA) followed by pairwise comparison using the Dunnett post-hoc test for THC-exposure vs. Veh. Non-normally distributed data were presented as box-plots (median as the middle line and 25/75% as box boundaries) and were statistically analyzed by Mann–Whitney U-test or by Kruskal–Wallis H-test followed by pairwise comparison using the Mann–Whitney U-test after Bonferroni correction (SPSS 24, SPSS Inc., Chicago, IL, USA). Differences associated with p ≤ 0.05 were considered statistically significant. Figures were created using the graphics program Corel Draw X5 (Corel Corporation, Ottawa, ON, Canada).









留言 (0)