
記住我
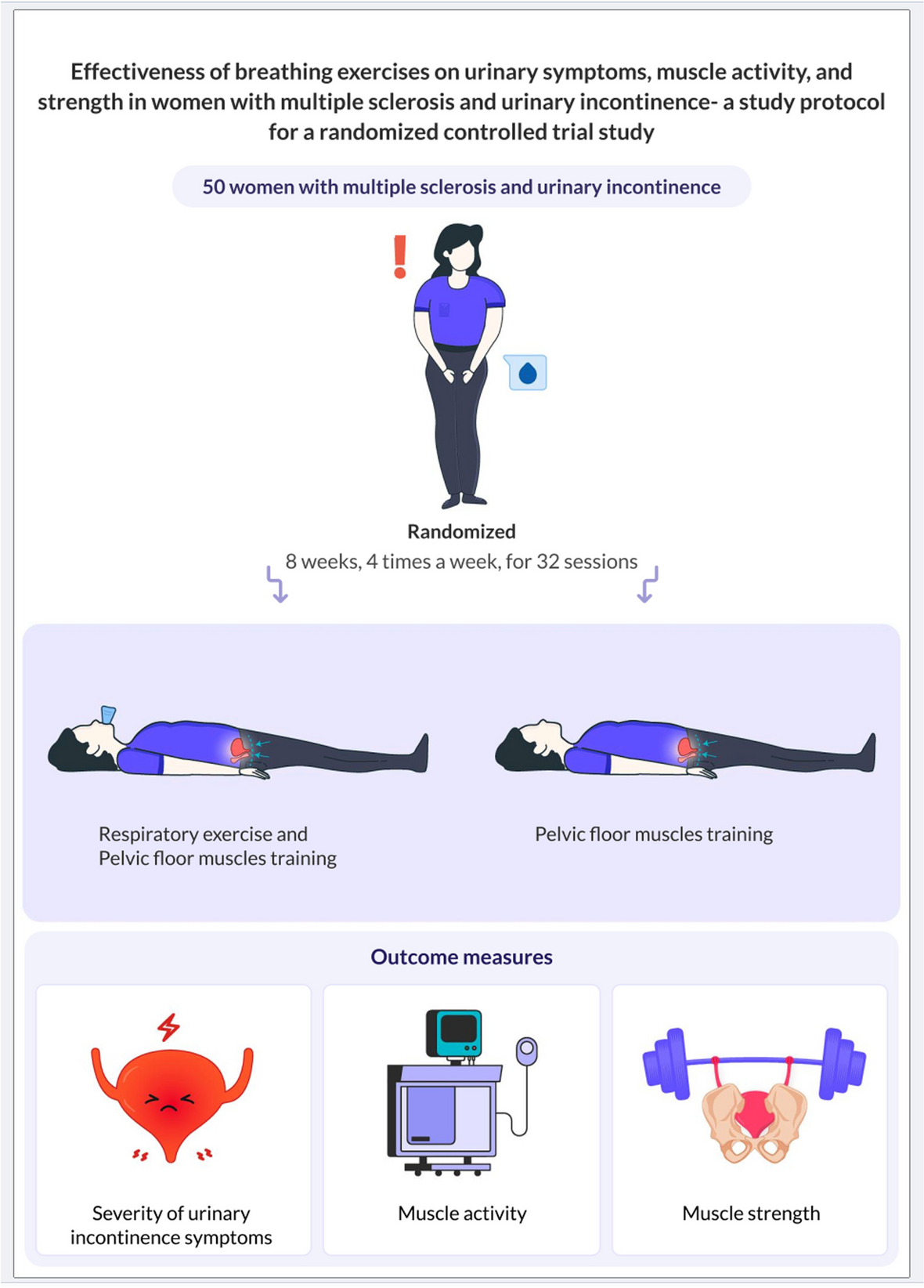

The primary aim of this RCT is to determine if, in people with MND who are referred for NIV, the proportion of participants using NIV for > 4 h/day during the intervention period is higher in the PSG (intervention) group than the control group. The primary hypothesis is that implementing NIV for people with MND with or at risk of chronic hypercapnic respiratory failure utilising PSG (intervention) to evaluate and titrate settings will lead to higher NIV usage compared to the control group. Usage is defined as using NIV for > 4 h/day while the participant is alive during the intervention period.
The secondary aims of this RCT are (i) to determine if total daily NIV use (hours/day) is greater in the PSG (intervention) group than in the control group; (ii) to evaluate whether the PSG group is superior to the control group in the change from baseline measures of gas exchange, respiratory function and patient-reported outcome measure (PROM) test scores as well as follow-up (review) physiological and objective sleep quality measures; and (iii) to examine differences in carer burden among people who care for participants in the PSG (intervention) and control group. Further exploratory aims are to determine whether particular patient-ventilator asynchrony sub-event types (ineffective efforts, double-trigger, multiple trigger) differ between the PSG and control group (RCT); to determine whether particular patient-ventilator asynchrony sub-type events are associated with objective NIV usage (RCT and 12-month cohort); and to examine associations between objective NIV use, survival and participant PROMs (RCT and 12-month cohort).
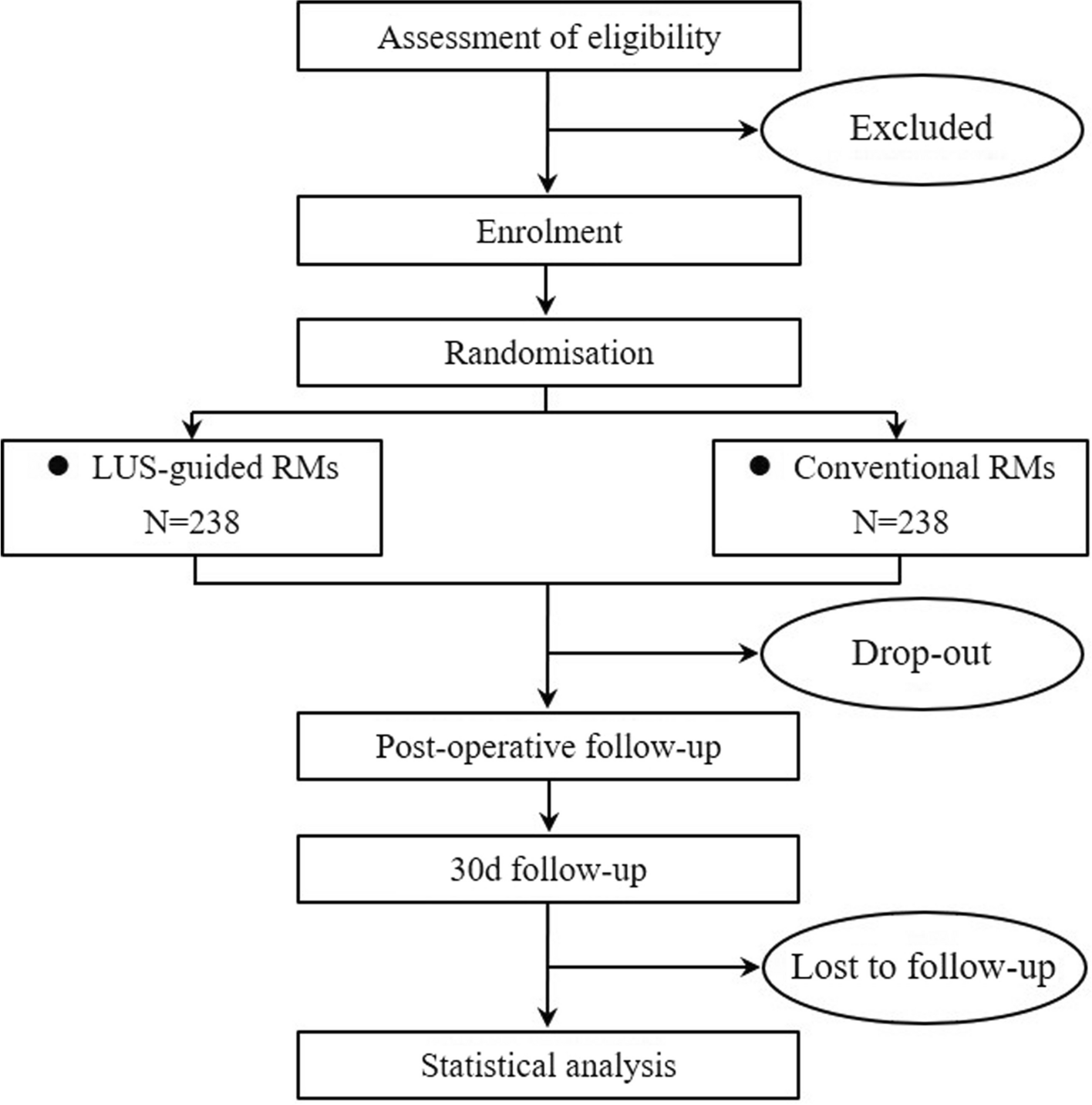
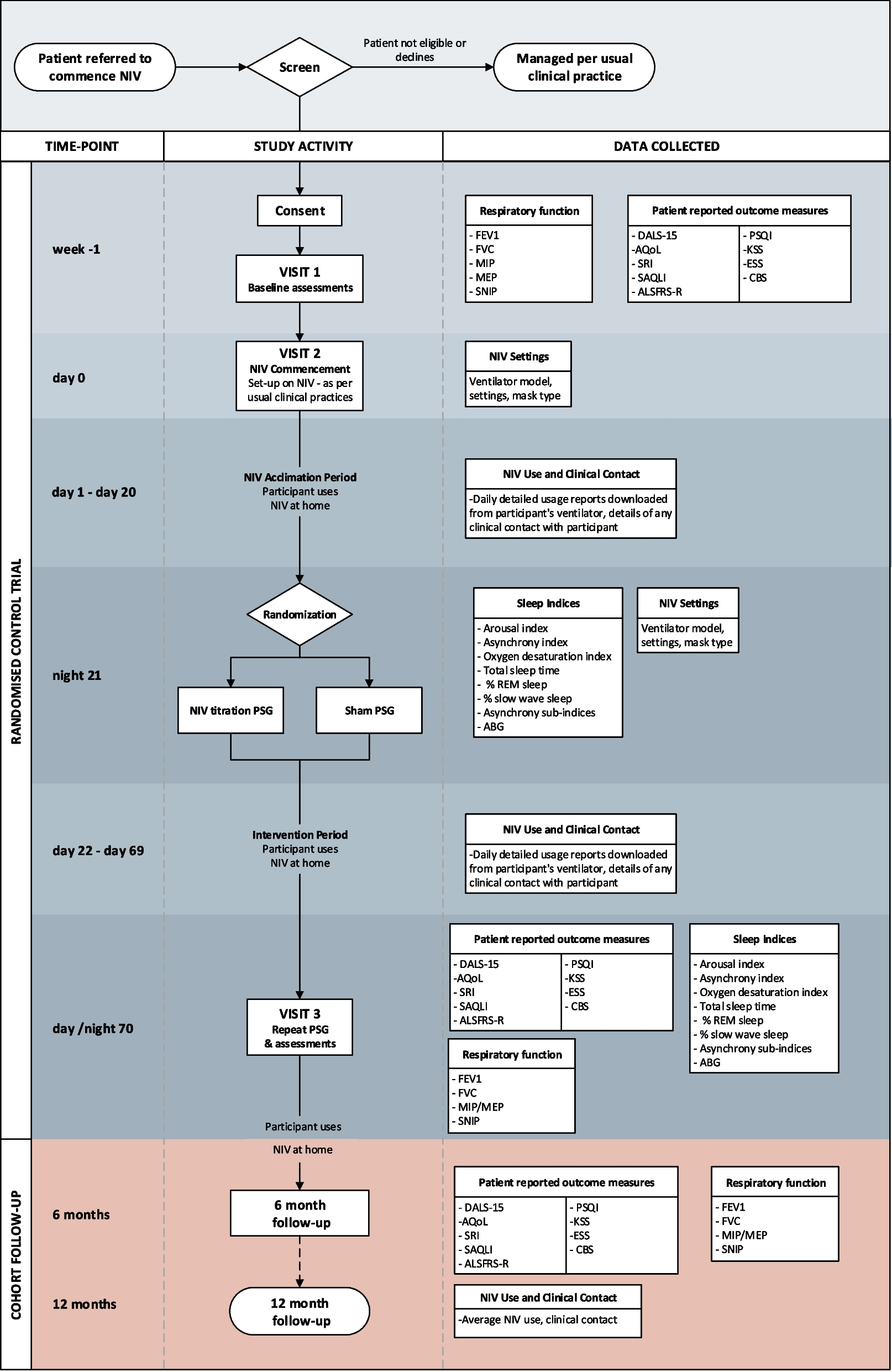
DesignThis trial of PSG-assisted commencement of NIV in MND follows similar methodology of the previous single-site trial [16]. The 3TLA trial is a two-arm, parallel group, superiority multi-centre trial conducted at MND care centres across Australia. These include Austin Health (Melbourne), Flinders Medical Centre (Adelaide), Macquarie University (Sydney), Royal Prince Alfred Hospital (Sydney), Sir Charles Gairdner Hospital (Perth), The Prince Charles Hospital (Brisbane) and Westmead Hospital (Sydney). Additional sites may be added as the trial progresses. The trial has been designed according to SPIRIT guidelines [19], will be conducted in accordance with the principles of Good Clinical Practice and reported according to the CONSORT and the complimentary StaRI standards for implementation studies [20]. Eligible participants will be randomised on a 1:1 basis to either the intervention group (daytime and PSG-assisted commencement of NIV settings; PSG) or control group (daytime commencement of NIV settings and sham PSG). The anticipated flow of participants through the trial is represented in Fig. 1.
Fig. 1
Participant flow through the 3TLA trial. NIV non-invasive ventilation, PSG polysomnography, PROMs patient-reported outcome measures
ParticipantsConsecutive individuals referred for implementation of NIV will be identified. Those who are interested in participating will be provided with a Participant Information and Consent Form by a member of the research team. Participants who meet the following inclusion criteria will be eligible to participate: (i) medically stable, (ii) suitable for outpatient implementation of NIV, (iii) clinical indication to commence long-term NIV according to local protocols, published guidelines [21, 22] and/or specialist opinion, (iv) confirmed clinical diagnosis of MND and (v) age > 18 years. Individuals will be excluded if they are (i) medically unstable, (ii) demonstrate hypoventilation attributable to medications with sedative/respiratory depressant side-effects, (iii) have previously used NIV for > 1 month in the previous 3 months, (iv) are unable to provide informed consent, (v) have experienced previous intolerance of NIV or (vi) are pregnant.
ProceduresVisit 1 (day 0) Baseline data collectionAll participants who meet the selection criteria and agree to participate will be invited to undertake visit 1. Baseline data collection will involve collection of written informed consent, screening data, general, respiratory and PROMs (listed below). Clinical measures (i.e. respiratory function) will be undertaken by the usual clinical staff at each site. Non-clinical measures (i.e. general information and PROMs) will be collected with the assistance of research staff. The specific measures collected are listed below.
General:
Demographic (age, sex, education level) and participant characteristics (height or arm span for non-ambulant [cm], weight [kg], medical history and current MND medications)
Respiratory function (or most recent results if undertaken in the preceding 2 weeks):
Lung function (spirometry measures of forced expiratory volume in 1 s [FEV1], forced vital capacity [FVC])
Maximal inspiratory/expiratory (MIP/MEP) and sniff nasal inspiratory pressure (SNIP)
PROMs:
Daytime sleepiness (Karolinska Sleepiness Scale [23] and the Epworth Sleepiness Scale [24])
Function (Amyotrophic Lateral Sclerosis Functional Rating Scale (Revised)) [25]
Dyspnoea (Dyspnoea Amyotrophic Lateral Sclerosis) [26]
Health-related quality of life (Assessment of Quality of Life [AoQL-8D] [27], Severe Respiratory Insufficiency Scale [28] and Calgary Sleep Apnoea Quality of Life Index [29])
Sleep quality (Pittsburgh Sleep Quality Index) [30]
Carer burden (Caregiver Burden Scale) [31]
Daytime NIV initiationParticipants in both groups will receive a standardised daytime commencement and titration of NIV performed by the same experienced clinical staff (respiratory physiotherapists, scientist, nurses or physicians, depending on site) who would normally undertake this task clinically. All sites in the proposed RCT are experienced providers of NIV in MND, practice according to current guidelines [21, 22, 32] and routinely address patient interface and NIV efficacy.
Daytime NIV commencement will involve individualising masking and NIV settings to subjectively optimise comfort, leak, patient-ventilator synchronisation, minute ventilation and oxygen saturation (SpO2). The NIV devices typically used clinically at each site will be utilised for this trial. These devices provide remote access or data extraction ensuring objective recording of adherence (usage). Devices will deliver bi-level NIV with a back-up respiratory rate. Rise time, inspiratory time, trigger and cycle sensitivities will be adjusted by the clinical staff as per usual practice.
Acclimatisation period (day 0–day 20)Following NIV commencement, participants will be sent home with the device and asked to wear it for a 3-week ‘acclimatisation’ period. During this time, the clinical team can adjust settings as deemed clinically necessary. Standard clinical procedures (e.g. phone calls and home visits) will not be restricted and will operate as per the usual process. Non-invasive ventilation usage data (hours/day) will be recorded throughout this period. Participants who cease NIV during the acclimatisation period will have their reasons for cessation recorded.
Visit 2—polysomnography and allocation (night 21) PolysomnographyAfter the 3-week acclimatisation period, all participants will return to the sleep laboratory for a full PSG as per standard procedures at each clinical site. A standard treatment or NIV PSG montage with additional device outputs will be provided to all sites for consistency. Participants will be set up by experienced sleep scientists. Full PSG will include standard electroencephalography, left and right electro-oculography, submental and diaphragm electromyography, measurement of airflow using the NIV device, body position, thoracic and abdominal respiratory effort bands and video recording. Oxygen saturations will be measured continuously with the use of a pulse oximeter and a finger probe. Measures of transcutaneous carbon dioxide (PtcCO2) will be performed using transcutaneous capnography with the probe heated to 43 °C. Once set up, an arterial blood gas (ABG) sample will be collected. Upon completion, the PSGs will be staged, scored [33] and reported using published criteria [34].
RandomisationA computer-generated randomisation list will be provided by an unblinded independent statistician, using randomly permuted blocks stratified by centre. The participants’ treatment allocation will be revealed by the sleep scientist or another unblinded member of the research team immediately before PSG commencement at visit 2. The allocation sequence will be concealed using a centralised trial database and will not be revealed to participants of either group or the clinical team. The unblinded individual will not be involved in data analyses or the clinical care of participants during the intervention period.
Intervention groupThe intervention group will undergo an attended overnight titration PSG. An experienced sleep scientist or registered nurse will adjust the NIV settings (and mask) using clinical experience and site-specific practices. In addition, each site will receive a standardised laboratory manual which they are able to refer to throughout the study period. A summary of instructions provided within the laboratory manual are outlined in Table 1.
Table 1 Summary of laboratory manual instructions on the titration of non-invasive ventilation provided to trial sitesIn the days following, the empirical NIV commencement settings will be revised based on the report recommendations from the PSG, as described under ‘Intervention period (day 22–day 69)’, below.
Control groupThe control group will undergo an attended overnight PSG without any titration to correct ventilator abnormalities. Masking issues (leak, noise, fit, etc.) identified by the participants in both arms will be addressed. In the control arm, however, potential issues observed during the PSG but not identified by participants will not be addressed [16]. This sham PSG thus controls for the effect of the PSG per se without contamination.
Intervention period (day 22–day 69)Following the PSG, a member of the research team, unblinded to group allocation, will then program the updated ventilator settings onto data cards, SD cards or USB sticks to be sent to participants with instructions on how to update their devices. Devices with a connection to cloud-based monitoring systems, such as AirView and Care Orchestrator, may have their settings updated using this software as appropriate, providing group allocation blinding is maintained.
Following the PSG, participants will enter the intervention period, during which there are no planned reviews or additional interventions. Participants will have access to their clinical teams as per standard procedures at each site. All contacts, interventions, adjustments to settings, changes to interfaces, etc. will be recorded and be included in the health economic analysis (the methodology of this will be reported separately). Non-invasive ventilation usage (hours/day) will be extracted via detailed reports from online platforms (e.g. AirView) or NIV data cards.
Visit 3—PSG2 and follow-up assessment (day/night 70)Following completion of the intervention period, participants will be asked to return to the study site for a follow-up assessment and second PSG; the follow-up assessment will mirror measures collected at baseline. Unattended home PSGs may be offered as an alternative to in-laboratory if clinically indicated. No settings will be adjusted during the PSG at visit 3. The sleep scientist performing analysis of these studies will be unaware of treatment allocation.
6- and 12-month cohort follow-upFollowing completion of the intervention period, participants will enter a 12-month open label cohort which will be reported separately from the RCT. Data collection timepoints within the cohort study will be conducted at 6 and at 12 months after the NIV initiation (visit 1). The cohort follow-up data collection will be undertaken within ( ±) a 2-week period of each designated timepoint and will be organised in accordance with usual clinic follow-up to minimise burden to participants and clinical teams. During each visit, survival and PROMs will be collected.
Data analysisA stand-alone statistical analysis plan will be finalised for the RCT before unblinding of the study database, detailing all analyses methods. Analyses will include randomised participants grouped according to their randomised intervention group. The primary estimand of clinical interest is the treatment effect of PSG (intervention) compared to control to titrate NIV therapy on usage of NIV (> 4 h/day) during the intervention period or until death (whichever occurs first), regardless of whether participants are prescribed a disease modifying medication, are non-adherent or discontinue NIV, experience adverse events or are admitted to hospital with an acute event during the intervention period. This estimand represents how patients are treated in clinical practice and assumes death is not a failure of the intervention under investigation.
For the primary outcome, we will use log-binomial regression adjusted for the average NIV usage per day during the acclimatisation period (in hours) to estimate the primary estimand, using control as the reference group. Secondary continuous outcomes will be analysed using linear regression adjusted for the baseline value, where applicable, with log transformation applied before fitting this model to outcomes that are positively skewed. Missing scale item data will be handled as per questionnaire specific recommendations. Missing data handling methods aligned with the estimand of primary interest, such as multiple imputation, will be used to handle missing values. Pre-specified subgroup analyses irrespective of the findings in the primary outcome will be performed to explore heterogeneity in the treatment effect for the subgroups defined by centres and MND phenotype including a main effect for subgroup and the interaction between subgroup and treatment group in the model. The analysis models for all outcomes will adjust for the randomisation stratification variable centre as a main effect.
Sample sizeFrom the single-site RCT [16] and data from an additional 56 people with MND receiving usual care at Austin Health (Melbourne), it was estimated that the proportion adherent for usual care (control group) was 56% (95% confidence interval [CI] 41 to 70). It was estimated by expert consensus that a 20% absolute difference between the treatment arms is meaningful. With 41% (i.e. lower bound of the 95% CI) in the control group and 61% in the PSG group (ratio of ≈1.49), the total sample size is 194 (two-sided significance level of 5%, power of 80%). Accounting for a conservatively estimated drop-out rate of 20%, the total required sample size is 244 (122 per arm).
Health economic evaluationCost-effectiveness analyses will be conducted alongside the trial from both a healthcare system and a patient/family perspective (including caregiver impacts). Additional extrapolation of the impact of NIV on survival will be incorporated. The model will be populated using outcomes data from the RCT and the subsequent cohort (NIV usage, health related quality of life (AQoL-8D) derived utility weights and survival). The costs of the intervention will be estimated from study protocols and budgets and the costs related to health resource use will be sourced from trial data, data linkage or self-report (hospitalisation, outpatient visits, disability supports and medication). A discount rate of 5% will be used for outcomes and costs incurred beyond 12 months. To access the effects of the uncertainty in estimates, a series of univariate sensitivity analysis will be conducted. Cost-effectiveness will be presented as a cost per additional hour of NIV use in the intervention group compared to the control group and cost per quality adjusted life year gained.
BlindingParticipants, the clinical team and research staff collecting outcome measures will all be blinded to group allocation. The sleep scientist/registered nurse supervising the PSG will be unblinded to group allocation. The clinical team and others blinded to the treatment allocation will not access the PSG, ABG or other data. The reporting sleep physicians at each site who will provide a treatment recommendation (according to their usual practice) for both groups will input data on a form that will not indicate the treatment allocation.
Trial managementData will be inputted into a secure, password-protected, web-based database Adept (https://adeptrs.com/). Any paper records will be kept in a secure filing cabinet at each site. Data will be de-identified at source. Polysomnography will be performed, scored and reported locally. The PSGs will also be de-identified and transferred to the central research team electronically for central reporting to allow for sensitivity analysis of reporting methods between sites. This process has successfully been used in previous trials conducted by members of the research team [16].
The Trial Management Group (TMG), comprising the chief investigator, trial manager, implementation science lead and central trial staff are responsible for the day-to-day delivery and conduct of the trial and liaise directly with trial staff at each site. The TMG group meet weekly or more frequently if required and report directly to the Trial Steering Committee (TSC).
The TSC, comprising co-chairs from the community support organisation partners, chief investigator, site principal investigators, trial manager and implementation science lead, meet quarterly to review trial progress, review recommendations from the Data and Safety Monitoring Board (DSMB) and agree on action plans to address issues as they arise.
Site principal investigators are responsible for all aspects of the day to day running of the trial at their sites including identification of potential participants and consent for trial participation. Each site has a dedicated trial coordinator responsible for coordination of participant visits, data collection, data entry and liaising with the central team.
Comprehensive monitoring and auditing procedures will be conducted by a trial monitor who is independent of the site investigators and sponsor. The trial monitoring plan involves six-monthly database audits and annual site visits to each trial site to audit trial procedures, including protocol adherence and data quality. In addition to the site visits, each site will provide a quarterly progress report to the Steering Committee.
The DSMB, consisting of a respiratory physician, neurologist, MND Australia nominee and biostatistician, meet biannually (or sooner as required) throughout the trial to advise the Steering Committee. The DSMB monitor recruitment, withdrawals and safety. All adverse events will be collected, recorded and assessed by the DSMB. Adverse events that have a material impact on the conduct of the research will be reported to the reviewing HREC in accordance with the NHMRC Position Statement: Monitoring and reporting of safety for clinical trials involving therapeutic products, November 2016.
The results of the trial will be published in peer-reviewed journals and presented at conferences and scientific meetings internationally. Additionally, the trial outcomes will be provided to trial participants in a summary and the MND community through patient support organisations.
留言 (0)