
記住我
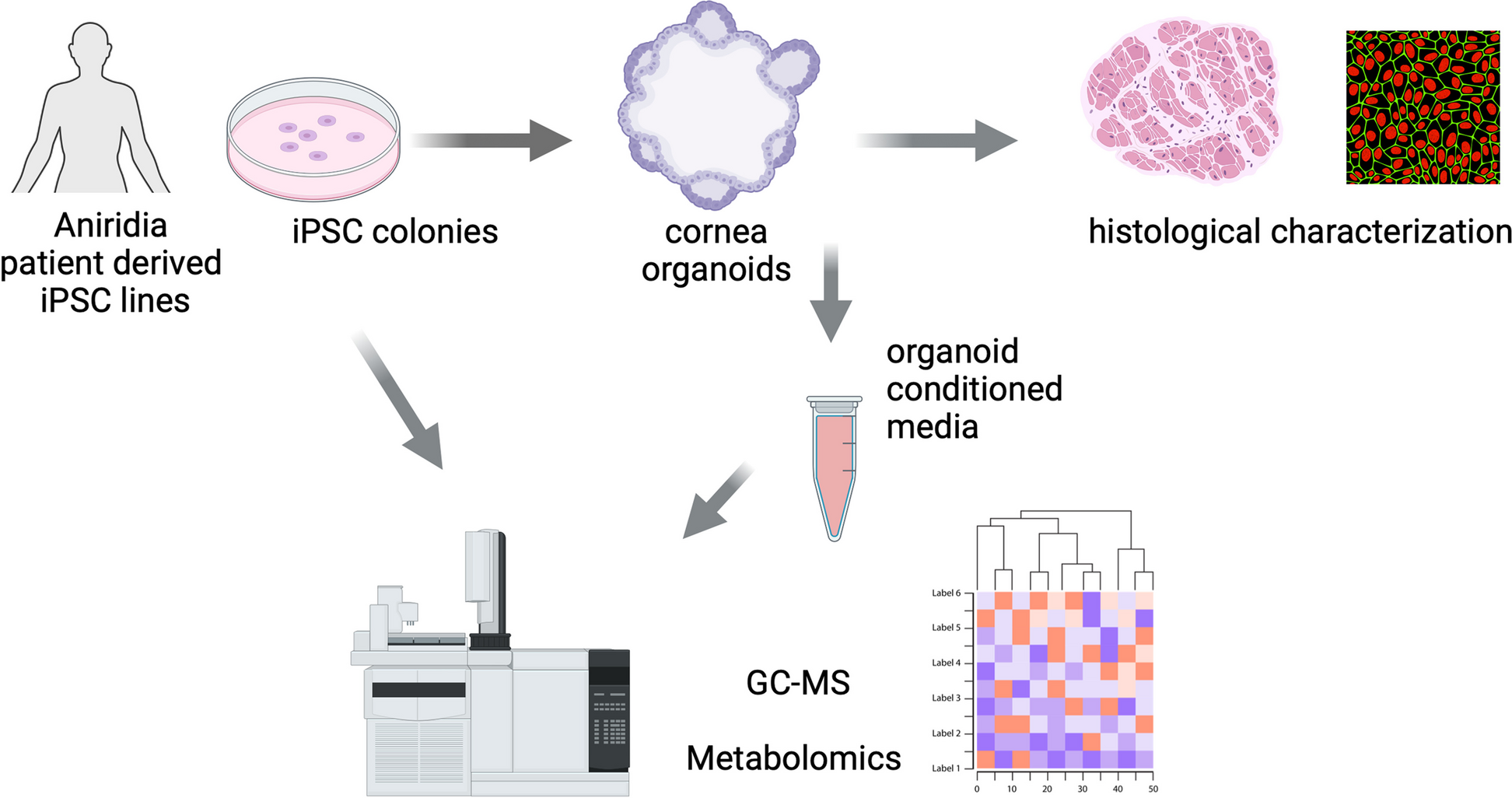






Patient-derived hiPSCs are valuable models for studying the pathophysiology underlying AAK in humans. In this study, we used three aniridia patient-derived hiPSCs lines with three different PAX6 heterozygous nonsense mutations and one wild-type (WT) hiPSC. hiPSCs were differentiated into cornea organoids and characterized their histologic and metabolomic profiles (Fig. 1).
Fig. 1
Schematic illustration of the study design
WT and patients-derived hiPSCs showed typical pluripotent stem cell morphologies with defined borders for pluripotent stem cells (Fig. 2A). Their pluripotency was also confirmed through pluripotency markers expression by OCT3/4 and NANOG immunofluorescence staining (Fig. 2B) and SOX2 and KLF4 mRNA levels by qPCR (Fig. 2C). Overall, the morphological characteristics, immunofluorescence staining, and qPCR analysis collectively demonstrate that the hiPSCs utilized in this study exhibit robust pluripotency. This confirms their suitability for modeling AAK and furthering our understanding of the disease mechanisms in a human context.
Fig. 2
Pluripotency of patients-derived and healthy control-derived hiPSCs. A) Brightfield light microscope images of hiPSCs (100 μM) B) Immunofluorescence staining of OCT3/4 and NANOG pluripotent stem cell markers (50 μM) C) mRNA expression levels of SOX2 and KLF4 pluripotent stem cell marker genes by qPCR
We next evaluated the metabolic profiles of four distinct hiPSC lines to further evaluate insight into the metabolic flux. Untargeted metabolomic analysis was conducted on undifferentiated cell samples using GC–MS. A total of 267 mass features were identified across all samples, with 90 of these being confirmed using reference databases (see Supplementary Excel file). The metabolomic profiles were subjected to multivariate statistical analysis, including principal component analysis (PCA), partial least squares discriminant analysis (PLS-DA), and a heatmap (Fig. 3A-D). The two-dimensional and three-dimensional PCA results in the absence of systemic errors or outliers in the dataset and demonstrated a clear contrast between the WT and AAK groups. The WT group (light blue) is observed to cluster separately from the AAK groups, indicating the presence of distinct metabolic profiles. Among the AAK groups, AAK-3 (green) exhibits some degree of overlap with WT, whereas AAK-1 (red) and AAK-2 (blue) are more distinctly separated. (Fig. 3A, B). The divergence between the WT and AAK groups is more pronounced in comparison to the PCA plot, which serves to illustrate the capacity of PLS-DA to differentiate the groups based on their metabolic profiles. A distinct separation between the metabolic profiles of the WT and AAK samples was evident in the PLS-DA models. The quality of the resulting PLS-DA model was assessed through internal cross-validation. The R2 and Q2 values were 0.99 and 0.77, respectively, indicating the presence of notable metabolic variations in the metabolome of the cell lines. (Fig. 3C). The metabolites that exhibited the greatest decrease in concentration between the WT and AAK samples were alanine, trans-3-hydroxy-L-proline, serine, glycine, succinic acid, leucine, 4-acetamidobutyric acid, glycerol 1-phosphate, and DL-glyceraldehyde. The concentrations of glyceraldehyde, cholesterol, threose, pantothenic acid, N-acetyl-L-aspartic acid, lysine, and asparagine are higher in WT samples, while other metabolites are lower significantly. This is in contrast to AAK samples, where the opposite was observed. The results of the hierarchical clustering analysis indicate that the samples derived from the WT cluster together, while those derived from AAK-1, AAK-2, and AAK-3 form separate clusters. (Fig. 3D).
Fig. 3
Metabolomic analysis of four different types of hiPSC cell lines. A) PCA score plot for metabolomic profiles of hiPSC cell lines at undifferentiated state B) 3D representation of PCA plot score C) PLS-DA score plot for metabolomic profiles of hiPSC cell lines at undifferentiated state (R2 = 0.99 and Q.2: 0.77) D) Heat map for cell metabolites (n = 3 biological replicate)
Generation of cornea organoids from aniridia patient-derived and healthy control-derived hiPSCsTo generate corneal organoids from hiPSCs, differentiation was initiated when the cells reached 70–80% confluency. For the first two days, the cells were exposed to differentiation medium (DM) to promote spontaneous differentiation. Following this, the cells were cultured in retinal differentiation medium (RDM) for differentiation into ocular cells. After 30 days of differentiation, the adherent cells were gently detached with a cell scraper and transferred to an ultra-low attachment six-well plate suspension culture. Over the next 45 days, the cells were maintained in corneal differentiation medium (CDM) to support the development and maturation of corneal-like transparent (Fig. 4A). Morphologically, the cornea organoids appeared as spherical structures with translucent or transparent layers (Fig. 4B), suggesting they are fluid-filled. Histological analysis using hematoxylin and eosin staining of cornea organoid sections displayed a stratified epithelium and stroma-like lumen within the semi-transparent or transparent-like regions (Fig. 4C), indicating a cellular organization that closely resembles that of the natural cornea. Further characterization of the cornea organoids was performed using immunofluorescence staining for Collagen IV, a corneal stroma protein [21, 59], and CK3, A corneal epithelial marker CK3 (Fig. 4D). Corneal organoids developed from hiPSCs showed significant positive expression of CK12, a biomarker of mature corneal epithelium (Fig. 4E) [49, 52, 64]. This finding suggests the successful differentiation of corneal epithelial cells and that our organoid model reflects the corneal epithelial phenotype. Keratocan, a proteoglycan specific to stromal cells [9, 30], and CK5, which is expressed in the basal layer of the corneal epithelium [46, 73], were immunofluorescence-stained in corneal organoids (Fig. 4F).
Fig. 4
Characterization of cornea organoids from aniridia patient-derived and healthy control-derived hiPSCs A) Schematic illustration of cornea organoid generation from hiPSCs B) Brightfield images of the corneal organoids (100 μm) C) Histological staining of corneal organoids with hematoxylin and eosin (100 μm) D) Immunofluorescence staining of Collagen IV and CK3 (50 μm). E) Immunofluorescence staining of CK12 (50 μm). F) Immunofluorescence staining of Keratocan and CK5 (50 μm). G) Immunofluorescence staining of Pax6 and p63 (50 μm). H) Immunofluorescence staining of Tuj1 (100 μm). I) Immunofluorescence staining of Ki67 (50 μm)
In our organoid model, CK5 expression facilitates precise epithelial cell feature mimicry. The possibility that organoids could mimic the corneal stroma is suggested by the expression of keratocan in their stromal-like layers. Pax6, the master regulator of eye development [25, 39], is also expressed in corneal epithelial cells. p63 also regulates eye development and epithelial stem cell maintenance and is highly expressed in limbal stem/progenitor cells [6, 51, 60]. Immunofluorescence staining for Pax6 and p63 showed that Pax6 expression was observed in corneal organoids developed from healthy (WT) hiPSCs, while no Pax6 expression was observed in organoids developed from Pax6 mutant hiPSCs. p63 expression was successfully expressed in the developed corneal organoids (Fig. 4G). To demonstrate the continued proliferation and development in corneal organoids, immunofluorescence staining of the cell proliferation marker Ki67 was performed (Fig. 4I) Nuclear localization of Ki67 in corneal organoids developed from healthy (WT) and Pax6 mutant hiPSC sources showed positive results. The combined positive results of limbal stem cell-associated protein p63 and Ki67 also support that the basic features of the corneal epithelium are mimicked. Tuj1 (β-III tubulin) positive expression was demonstrated in the developed corneal organoids (Fig. 4H). Tuj1 supports the presence of neuroectodermal cells that simulate the development of corneal innervation. Tuj1 is known as a marker of mature and developing neuronal cells and is critical for the demonstration of a phenotype that supports healthy innervation processes in corneal tissue [5, 28]. The distribution of Tuj1 positive cells by Tuj1 immunofluorescence staining supports the model representing the early stages of corneal innervation. These findings demonstrate that corneal organoids from both patients-derived and healthy control-derived hiPSCs can successfully mimic the structural organization of the natural cornea.
Metabolomic profiles of cornea organoids from aniridia patient-derived and healthy control-derived hiPSCsFollowing the initiation of the differentiation process at days 30, 44 and 60, the metabolomic profiles of organoids are elucidated through the application of untargeted GC–MS metabolomic analysis. A total of 540 mass features were identified across all samples, with 134 of these being confirmed using reference databases (see Supplementary Excel file). A multivariate statistical analysis was conducted on the metabolomic profiles using partial least squares discriminant analysis (PLS-DA) heatmaps, as illustrated in Fig. 5A-F. The metabolomic profiles of AAK-1 and AAK-2 organoids were similar, while those of AAK-3 and WT were distinct at day 30 (Fig. 5A). At day 44, the groups were combined, and a similar metabolomic profile was observed between them, with the exception of AAK-1 (Fig. 5B). Finally, at day 60, AAK-1 and AAK-2 organoids exhibited a metabolomic profile that clustered together, while AAK-3 and WT organoids displayed a distinct profile (Fig. 5C). The heatmap for day 30 illustrates the comparative concentrations of diverse metabolites across the various experimental groups. Metabolites such as D-Ala/D-Ala, L-proline, and capric acid displayed distinctive patterns between the WT and AAK groups. The results of the hierarchical clustering analysis indicated that the samples from the WT group clustered together, while the samples from the AAK-1, AAK-2, and AAK-3 groups formed separate clusters (Fig. 5D). The heatmap for day 44 demonstrated a more pronounced differentiation in metabolite concentrations in comparison to day 30. Notably, metabolites such as D-Ala/D-Ala, L-proline, and xylitol exhibit pronounced differences between the WT and AAK groups. The clustering pattern remained consistent, with the WT samples forming a distinct cluster from the AAK groups (Fig. 5E). The day 60 heatmap demonstrated the existence of discrete metabolic profiles for the WT and AAK groups. Metabolites such as beta-glycerolphosphate, tartaric acid, and capric acid exhibited notable distinctions. The results of hierarchical clustering indicated the presence of discrete clusters for the WT and AAK groups, which is consistent with the findings of previous heatmaps (Fig. 5F).
Fig. 5
Exometabolomic analysis of three patient-derived and healthy control-derived cornea organoids. A) PLS-DA score plot for exometabolomic profile of AAK and WT organoids at day 30 (R2 = 0.98 and Q2: 0.33) B) PLS-DA score plot for exometabolomic profile of AAK and WT organoids at day 44 (R2 = 0.76 and Q2: 0.06) C) PLS-DA score plot for exometabolomic profile of AAK and WT organoids at day 60 (R2 = 0.93 and Q.2: 0.63) D) Group average heat map for cell culture medium metabolites from AAK and WT Organoids at day 30 (n = 3 biological replicate for each group) E) Group average heat map for cell culture medium metabolites from AAK and WT Organoids at day 44 (n = 3 biological replicate for each group) F) Group average heat map for cell culture medium metabolites from AAK and WT Organoids at day 60 (n = 3 biological replicate for each group)
We further performed the comparison between the groups Metabolite Set Enrichment Analysis (MSEA) in metabolic pathways. AAK-1 and AAK-2 revealed a notable enrichment in pathways including alanine, aspartate, and glutamate metabolism; glyoxylate and dicarboxylate metabolism; glycolysis/gluconeogenesis; and starch and sucrose metabolism. Among these, alanine, aspartate, and glutamate metabolism exhibited the highest degree of enrichment, indicating substantial metabolic differences between AAK-1 and AAK-2 (Fig. 6A). Similarly, the comparison between AAK-1 and AAK-3 revealed significant enrichment in alanine, aspartate, and glutamate metabolism, along with glyoxylate and dicarboxylate metabolism, glycolysis/gluconeogenesis, and the citrate cycle (TCA cycle). The alanine, aspartate, and glutamate metabolism pathways exhibited the greatest degree of enrichment, indicating that it is subject to significant metabolic alterations between AAK-1 and AAK-3 (Fig. 6B). The comparison between AAK-2 and AAK-3 also revealed significant enrichment in alanine, aspartate, and glutamate metabolism, along with glyoxylate and dicarboxylate metabolism, glycolysis/gluconeogenesis, and starch and sucrose metabolism. This consistent enrichment highlights the existence of significant metabolic differences between AAK-2 and AAK-3 at D30 (Fig. 6C).
Fig. 6
The Metabolite Set Enrichment Analysis (MSEA) in metabolic pathways between various AAK organoids at different time points. A) AAK-1 vs. AAK-2 at day 30 B) AAK-1 vs. AAK-3 at day 30 C) AAK-2 vs. AAK-3 at day 30 D) AAK-1 vs. AAK-2 at day 44 E) AAK-1 vs. AAK-3 at day 44 F) AAK-2 vs. AAK-3 at day 44 G) AAK-1 vs. AAK-2 at day 60 H) AAK-1 vs. AAK-3 at day 60 I) AAK-2 vs. AAK-3 at day 60. The y-axis lists the 25 most significantly enriched metabolic pathways, while the x-axis represents the -log10 (p-value) of each pathway. The size of the dots is proportional to the enrichment ratio, and the color gradient from yellow to red indicates the p-value, with red representing the most significant enrichment
A comparison between AAK-1 and AAK-2 at D44 revealed a notable enrichment in pathways including valine, leucine, and isoleucine biosynthesis; alanine, aspartate, and glutamate metabolism; glyoxylate and dicarboxylate metabolism; and the TCA cycle. The valine, leucine, and isoleucine biosynthesis pathways exhibited a pronounced increase in enrichment, underscoring significant metabolic disruptions between AAK-1 and AAK-2 (Fig. 6D). A similar pattern of enrichment was observed in the comparison between AAK-1 and AAK-3, with an increase in the valine, leucine, and isoleucine biosynthesis pathway, alanine, aspartate, and glutamate metabolism, glyoxylate and dicarboxylate metabolism, and the TCA cycle. This consistent pattern of enrichment underscored the existence of substantial metabolic alterations between AAK-1 and AAK-3 at D44 (Fig. 6E). In the comparison between AAK-2 and AAK-3, there was a notable enrichment in pathways including valine, leucine, and isoleucine biosynthesis; alanine, aspartate, and glutamate metabolism; glyoxylate and dicarboxylate metabolism; and the TCA cycle. These findings indicate that there are persistent metabolic differences between AAK-2 and AAK-3 at this time point (Fig. 6F).
At D60, the comparison between AAK-1 and AAK-2 continued to demonstrate enrichment in valine, leucine, and isoleucine biosynthesis; alanine, aspartate, and glutamate metabolism; glyoxylate and dicarboxylate metabolism; and the TCA cycle. This consistent pattern of enrichment suggests the presence of ongoing metabolic differences between AAK-1 and AAK-2 (Fig. 6G). The comparison between AAK-1 and AAK-3 also revealed persistent enrichment in these pathways, indicating significant metabolic disruptions between these strains at D60 (Fig. 6H). Finally, the comparison between AAK-2 and AAK-3 demonstrated sustained enrichment in valine, leucine, and isoleucine biosynthesis, alanine, aspartate, and glutamate metabolism, glyoxylate and dicarboxylate metabolism, and the TCA cycle. These findings underscore the persistence of metabolic disparities between AAK-2 and AAK-3 at D60 (Fig. 6I). Overall, the metabolite enrichment analysis across diverse AAK organoids and time points consistently demonstrated substantial alterations in pivotal metabolic pathways, notably those pertaining to amino acid metabolism and primary energy production. These outcomes offer invaluable insights into the metabolic consequences of different AAK organoids.
MSEA revealed the existence of enriched pathways associated with upregulated metabolites, including amino acid biosynthesis (e.g., L-serine, L-methionine, glutamic acid), carbohydrate metabolism (e.g., L-sorbose, cellobiose), nucleotide metabolism (e.g., uracil, thymine), and lipid metabolism (e.g., heptadecanoic acid, methyl linolenate). (Fig. 7A, C, E) In contrast, the downregulated metabolites were found to be enriched in pathways related to amino acid degradation (e.g., sarcosine, urea), lipid metabolism (e.g., myristic acid, methyl palmitoleate), and energy production (e.g., fumaric acid, pyruvic acid). The results demonstrate extensive metabolic alterations across the AAK mutants, with both shared and distinct metabolomic profiles indicating the specific functions of AAK-1, AAK-2, and AAK-3 in regulating amino acid metabolism, carbohydrate utilization, and energy balance (Fig. 7B, D, F).
Fig. 7
The Metabolite Set Enrichment Analysis (MSEA) in metabolic pathways between various AAK organoids at day 60. A) WT vs. AAK-1 upregulated B) WT vs. AAK-1 downregulated C) WT vs. AAK-2 upregulated D) WT vs. AAK-2 downregulated E) WT vs. AAK-3 upregulated F) WT vs. AAK-3 downregulated G) Proportional Venn diagram for the upregulated metabolites between WT vs AAK-1, WT vs AAK-2 and WT vs AAK-3 H) Proportional Venn diagram for the downregulated metabolites between WT vs AAK-1, WT vs AAK-2 and WT vs AAK-3. The y-axis lists the 25 most significantly enriched metabolic pathways, while the x-axis represents the -log10 (p-value) of each pathway. The size of the dots is proportional to the enrichment ratio, and the color gradient from yellow to red indicates the p-value, with red representing the most significant enrichment
The metabolomic analysis revealed significant alterations in both upregulated and downregulated metabolites across the WT and AAK groups. Sixteen metabolites were commonly upregulated across all groups (WT vs. AAK-1, WT vs. AAK-2, and WT vs. AAK-3). These included diethyl oxalpropionate, L-methionine, uracil, aspartic acid, tryptophane, xanthosine, thymine, and phenyl-beta-glucopyranoside. These include ide, phenylalanine, sedoheptulose anhydride monohydrate, alpha-D-glucosamine 1-phosphate, 1-methylnicotinamide, cysteine, gluconic acid, L-pyroglutamic acid, and citric acid, which collectively reflect shared disruptions in amino acid biosynthesis, nucleotide metabolism, and carbohydrate pathways. A comparison of the WT vs. AAK-1 and WT vs. AAK-2 groups revealed that nine metabolites were upregulated. The presence of L-sorbose, 2-keto-L-gulonic acid, trans-4-hydroxy-L-proline, L-glutamic acid, maltose, cellobiose, tyrosine, glucoheptonic acid, and 5-aminovaleric acid indicates a common increase in amino acid and sugar metabolism. A comparison of WT vs. AAK-1 and WT vs. AAK-3 revealed three shared upregulated metabolites: sucrose, malonic acid, and 2-amino-1-phenylethanol. This suggests alterations in energy intermediates. A comparison of WT vs. AAK-2 and WT vs. AAK-3 revealed nine metabolites, including L-glutamine, L-serine, lysine, L-proline, putrescine, cycloleucine, 2-amino-2-methyl-1,3-propanediol, glycine, and glycerol 1-phosphate, that were consistently upregulated. This finding reflects an increase in amino acid biosynthesis and nitrogen metabolism. In the set of unique upregulated metabolites, WT vs. AAK-1 exhibited six: pipecolic acid, 2-ketoisocaproic acid, DL-3-aminoisobutyric acid, galacturonic acid, heptadecanoic acid, and lactamide. The WT vs. AAK-2 comparison exhibited a unique upregulation of isoleucine, L-cysteine, L-leucine, and talose, which suggests the regulation of branched-chain amino acids. In the WT vs. AAK-3 comparison, L-alanine, resveratrol, serine, D-lyxosylamine, L-homoserine, glycolic acid, D-Ala-D-Ala, lactobionic acid, and tartaric acid were uniquely upregulated, indicating the involvement of pathways associated with antioxidants and sugar metabolism (Fig. 7G). Four metabolites (sarcosine, serine, carbamic acid ethyl ester (urethane), and glutamic acid) exhibited consistent downregulation across WT vs. AAK-1, WT vs. AAK-2, and WT vs. AAK-3, indicating disruptions in amino acid degradation and nitrogen metabolism. In comparison between WT and AAK-1, urea was the sole metabolite that exhibited a unique downregulation, indicating a potential reduction in urea cycle activity. A comparison between WT vs AAK-1 and WT vs AAK-2 revealed that eleven metabolites were shared between the two groups and exhibited a downregulation: 3-methyl-2-oxobutanoic acid, D-lyxose, myristic acid, acetol, L-valine, threose, pyruvic acid, N-ethylglycine, DL-3-aminoisobutyric acid, pipecolic acid, and ribitol. This finding reflects impaired lipid catabolism and sugar processing. The unique downregulated metabolites in WT vs. AAK-1 included glycerol, fumaric acid, glyceric acid, phosphoric acid, acetylisatin, cis-4-hydroxycyclohexanecarboxylic acid, 2-piperidone, allo-inositol, N-methyl-DL-glutamic acid, and D-threitol, indicating disturbances in phosphate and energy metabolism. In the WT vs. AAK-2 comparison, twelve unique downregulated metabolites were identified. The presence of D-mannitol, hypotaurine, methyl linolenate, L-norleucine, DL-isoleucine, methyl palmitoleate, DL-glyceraldehyde, threonine, tyrosine, phenylacetaldehyde, iminodiacetic acid, and D-allose suggests disruptions in lipid and sugar pathways. In WT vs. AAK-3, the unique downregulated metabolites included L-homoserine, 1-methylhydantoin, glycolic acid, putrescine, methyl-beta-D-galactopyranoside, and resveratrol, which further highlight disruptions in polyamine biosynthesis, antioxidant pathways, and sugar metabolism (Fig. 7H).
留言 (0)