
記住我


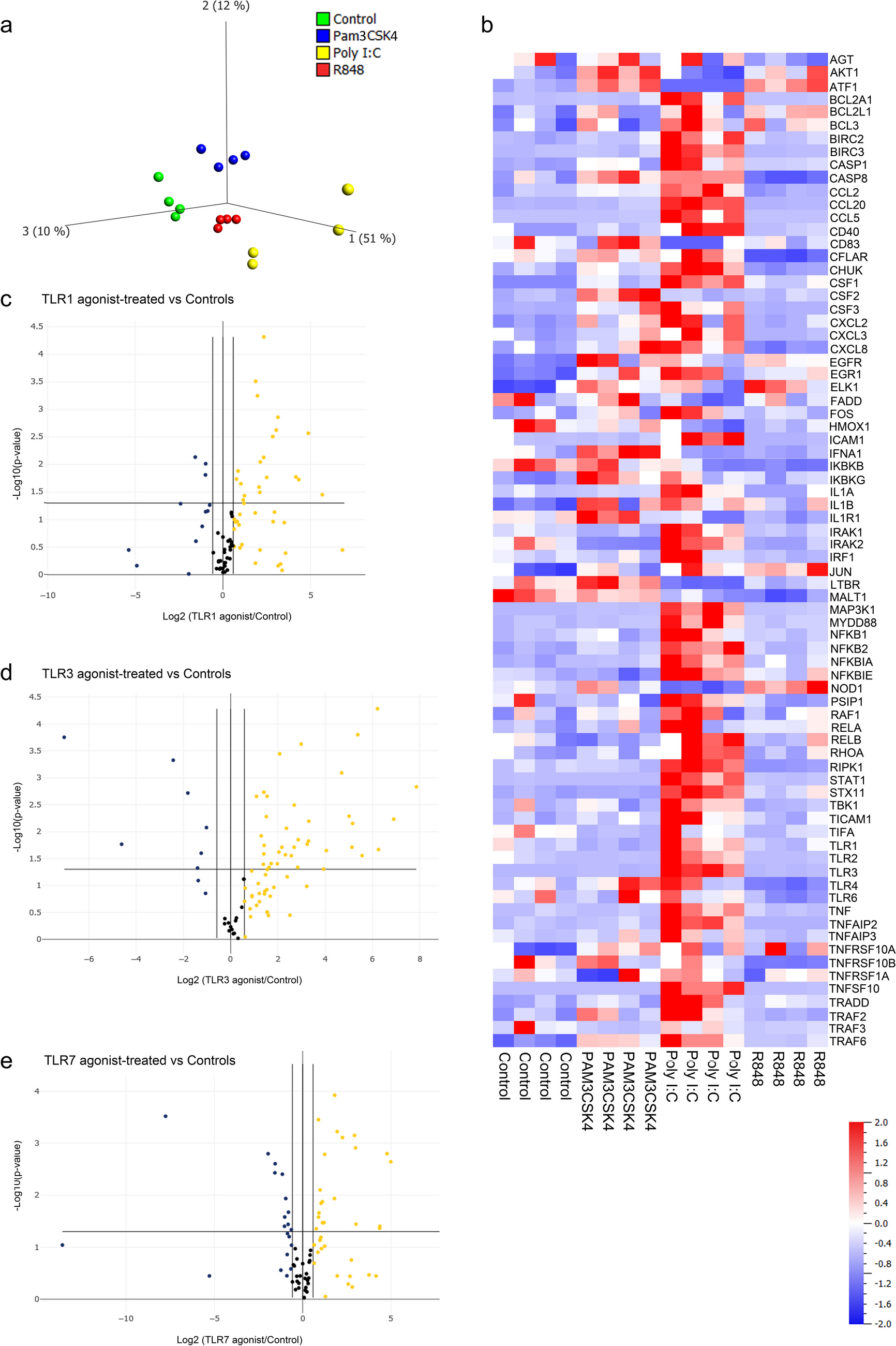
To elucidate potential alterations of RLRs in COPD, we initially reanalyzed a previously published scRNA-seq dataset (GSE171541) using the Seurat approach, which included three normal and six COPD lung tissue samples. After quality control filtering, a total of 27,062 unique genes were obtained from more than 66,000 cells distributed among nine lung tissue samples. We identified a total of 15 distinct cell clusters based on raw cell annotations after normalizing gene expression (Fig. 1A), including 9 immune cells [B cells (1211 cells), dendritic cells (DCs, 4772 cells), macrophages (21,006 cells), mast cells (2197 cells), monocytes (1637 cells), natural killer cells (NKs, 7968 cells), neutrophils (778 cells), proliferating cells (797 cells) and T cells (6933 cells)]; and six parenchymal cells [alveolar type 1 cells (AT1, 1358 cells), alveolar type 2 cells (AT2, 7476 cells), ciliated cells (2156 cells), club cells (2156 cells), endothelial cells (3171 cells) and stromal cells (2298 cells)]. Of these, approximately 44,927 cells belonged to control samples, while 21,681 cells were from COPD patients. The proportions of different cell types in each sample revealed that neutrophils, mast cells, and AT1 cells were abundant in COPD (Fig. 1B). In contrast, T cells, stromal cells, proliferating cells, NKs, monocytes, macrophages, endothelial cells, DCs, club cells, B cells and AT2 cells were abundant in control samples. Figure 1C shows the expression of the top 5 characterized genes in each cell.
Fig. 1
Identification of cell clusters in COPD by scRNA-seq data. (A) tSNE clustering of single cells from 3 control and 6 COPD lung tissue samples from the scRNA-seq dataset GSE171541, illustrating the formation of 15 major clusters; each point represents a cell, with each color corresponding to a different cell cluster. (B) Stacked bar charts display the proportion of each cell type in lung tissues of control and COPD groups. (C) Heatmap shows the top 5 unique feature genes in each annotated cell cluster. (D) Chord diagrams illustrate the differences in the number (left) and intensity (right) of interactions within the intercellular communication networks between control and COPD groups. Thicker lines indicate stronger interactions, with red or blue denoting an increase or decrease in signaling pathways in the COPD group compared to the control group, respectively. (E) Bar charts highlight the differences in intercellular pathways between control and COPD groups. Abbreviation: COPD: chronic obstructive pulmonary disease; scRNA-seq: single-cell RNA sequencing
Subsequently, we performed CellChat analysis to explore cell-to-cell interactions during COPD progression. The number and intensity of interactions were enhanced from normal to COPD. In particular, macrophages, DCs and AT2 cells in the COPD group exhibited a greater number of interactions and interaction strength with other cell types relative to the control group. In contrast, monocytes and proliferating cells communicated frequently in the control group (Fig. 1D). Finally specific pathways were identified between the control and COPD groups by comparing the interaction strength of each pathway (Fig. 1E). MSTN, PERIOSTIN, OPIOID, EPO, and BAFF signaling pathways were particularly active in COPD patients, whereas control individuals exhibited relatively high PRL, FLT3, NGF, MIF and TNF signaling pathway activity.
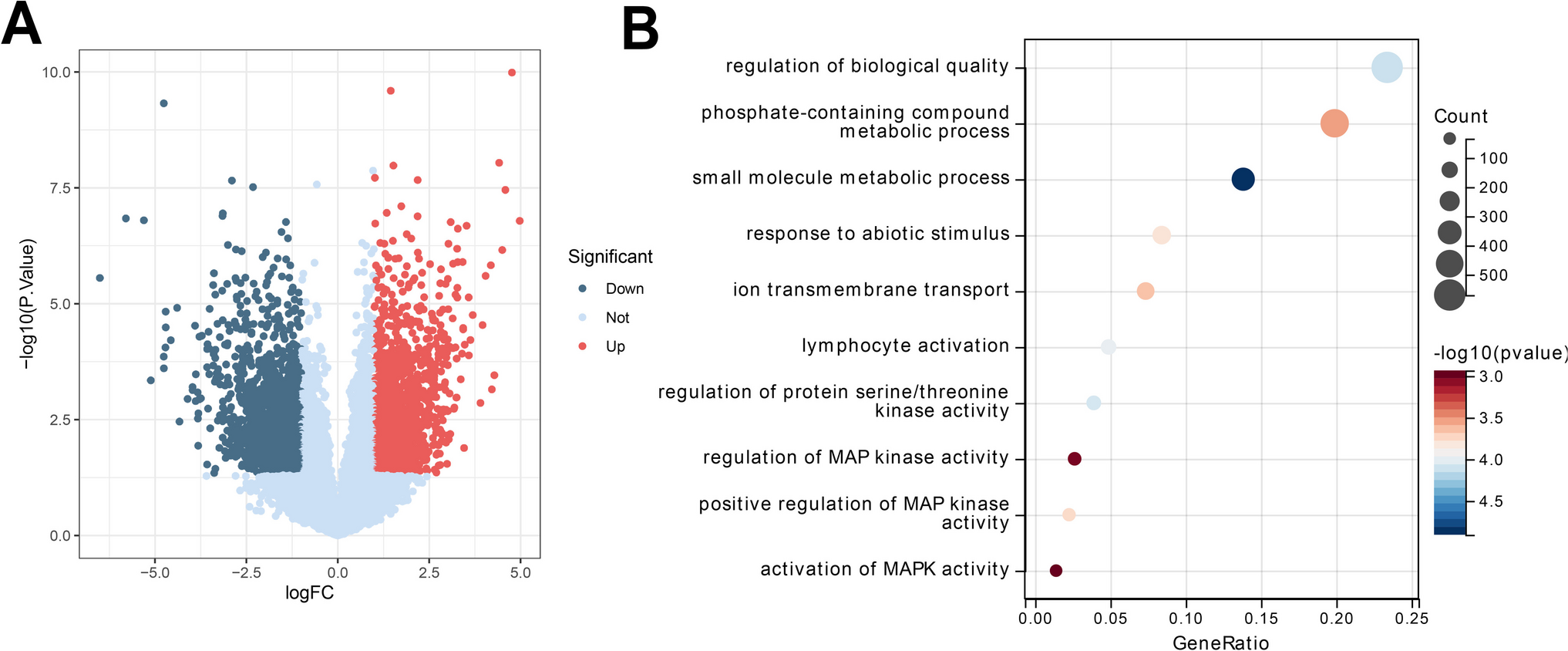
Screening RLRs-Related DEGsThe FidAllMarkers function was performed to identify a total of 2896 DEGs between COPD and normal samples (Additional file 1: Table S2), with an adjusted P-value < 0.05 and |log2 FC|> 1. Meanwhile, we identified 93 COPD-associated RLRs (Additional file 1: Table S3) by intersecting 2896 DEGs with 1185 RLRs (Fig. 2A). Gene ontology (GO) enrichment analysis showed that these 93 COPD-associated RLRs were involved in the regulation of chromosome segregation, spindle organization, TAP complex, spindle, ATP hydrolysis activity and MHC protein binding (Fig. 2B). The KEGG pathway enrichment analysis showed that these differentially expressed COPD-associated RLRs mainly affected the C-type lectin receptor signaling pathway, Epstein-Barr virus infection, Viral carcinogenesis, Human immunodeficiency virus 1 infection and other pathways (Fig. 2C).
Fig. 2
Identification of COPD-related RLRs based on scRNA-seq Data. (A) Venn diagram showing the intersection of COPD-related DEGs with 1185 RLRs. (B) GO enrichment analysis of 93 COPD-related RLRs. (C) KEGG pathway enrichment analysis of 93 COPD-related RLRs. (D-G) tSNE plots depicting the distribution of RLRs in cells based on the AUCell (D), UCell (E), singscore (F), and ssGSEA (G) algorithms. Abbreviation: COPD: chronic obstructive pulmonary disease; RLRs: R-loop regulators; scRNA-seq: single-cell RNA sequencing
We subsequently focused on the relationship between COPD-derived cells and COPD-associated RLRs. Based on the scRNA-seq expression matrix, we used multiple scoring algorithms to quantify the R-loop score in COPD cells based on the expression of RLRs, and employed algorithms to depict the distribution of RLRs in single cells of lung tissues of the control and COPD groups (Fig. 2D-G). It was found that the R-loop score of COPD patients was significantly lower than that of the control group in the AUCell, UCell, singscore and ssGSEA algorithms (Fig. 3A-D). Similarly, in the single-cell validation dataset GSE173896, R-loop scores for COPD patients, calculated using the AUCell, UCell, singscore, and ssGSEA algorithms, were significantly lower compared to the control group (Additional file 2: Fig. S1). Therefore, we further evaluated the R-loop scores in various cells between the two groups in both the test and validation single-cell sets. The results revealed that, in both sets, neutrophils from the COPD group displayed significantly reduced R-loop scores across all four algorithms compared to the control group (Fig. 3E-H and Fig. S1E-H). These findings suggest that a reduced R-loop score may predict the development of COPD, with neutrophils being the cells most closely associated with this decrease.
Fig. 3
Distribution of R-loop scores based on RLRs in COPD. (A-D) Violin plots comparing R-loop scores between control and COPD groups based on AUCell (A), UCell (B), singscore (C), and ssGSEA (D) algorithms. (E–H) Violin plots illustrating the differences in R-loop scores among different cell subtypes based on AUCell (E), UCell (F), singscore (G), and ssGSEA (H) algorithms. Compared with the control groups, * P < 0.05; ** P < 0.01; *** P < 0.001. Abbreviation: COPD: chronic obstructive pulmonary disease; RLRs: R-loop regulators
Assessment of Intercellular Communication and Prediction of Transcription Factor Activity among Patients with Different R-loop RisksUsing the R-loop scores constructed via the ssGSEA method, COPD patients are classified into high-risk (low R-loop score) and low-risk (high R-loop score) COPD groups. We further assessed intercellular communication and transcription factor activity between high-risk (low R-loop score) and low-risk (high R-loop score) COPD groups. Firstly, we assessed the proportions of different cell types between the two groups (Fig. 4A), revealing that AT1 cells and neutrophils were abundant in the high-risk COPD (low R-loop score) groups, whereas macrophages and proliferating cells were abundant in the low-risk (high R-loop score) COPD groups. Secondly, we constructed aggregated intercellular communication networks based on the number of interactions and interaction weights (Fig. 4B), and plotted the interaction strengths of cells transmitting and receiving signals (Fig. 4C-D). Macrophages were found to be important cells for intercellular communication in the low-risk (high R-loop score) COPD groups, whereas NKs played an important role in intercellular communication in the high-risk (low R-loop score) COPD groups. Finally, transcription factor analysis was performed. Figure 4E shows the top 10 most active TFs in both groups, including PRDM5, RNF114, RFX3, ARID3A, E2F8, E2F1, MYBL2, KLF5, XBP1, and ZNF274 in the high-risk (low R-loop score) COPD groups; and TFEC, IRF8, USF2, SPL1, BHLHE41, MLX, MAFF, CEBPB, MAFB, and JUNB in the low-risk (high R-loop score) COPD groups. Figure 4F shows the differences in transcriptional regulator activity between the two groups.
Fig. 4
Evaluation of intercellular communication and TFs activity in high-risk and low-risk COPD groups based on scRNA-seq data. (A) Stacked bar charts display the proportion of each cell type in lung tissues of high-risk and low-risk COPD groups. (B) Chord diagrams illustrate the differences in the number (left) and intensity (right) of interactions within the intercellular communication networks between high-risk and low-risk COPD groups. Thicker lines indicate stronger interactions, with red or blue denoting an increase or decrease in signaling pathways in high-risk and low-risk COPD groups. (C) Bar charts show the relative strength of interactions among incoming signals in different cell types between high-risk and low-risk COPD groups. (D) Bar charts show the relative strength of interactions among outgoing signals in different cell types between high-risk and low-risk COPD groups. (E) Top 10 active TFs regulating in high-risk and low-risk COPD groups. (F) Heatmap visualizes the regulation of TFs in high-risk and low-risk COPD groups. Abbreviation: COPD: chronic obstructive pulmonary disease; TFs: transcription factor; scRNA-seq: single-cell RNA sequencing
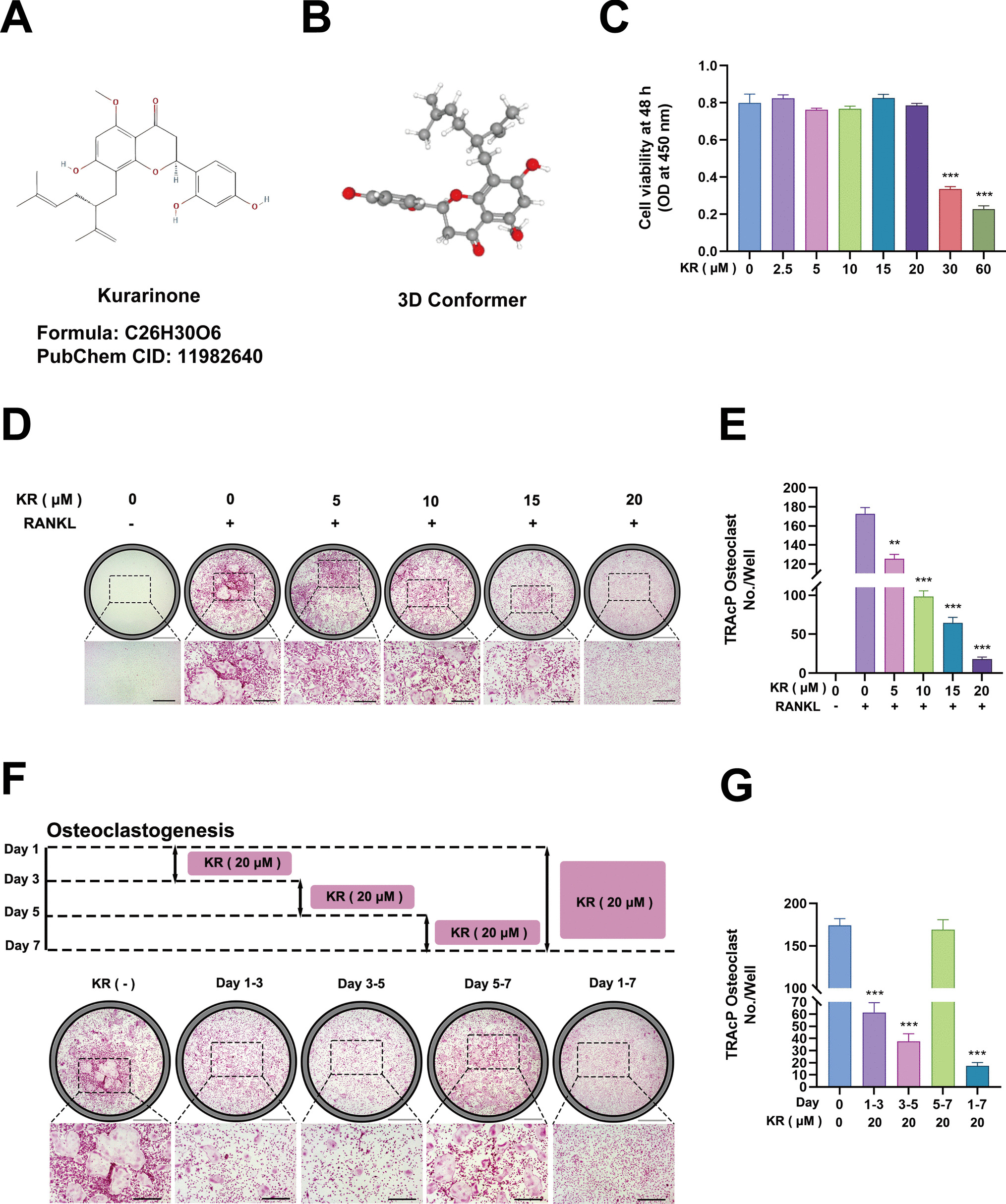
Machine Learning Algorithms IdEntify Hub RLRsTo further leverage the transcriptional features we developed, we extracted 93 RLRs identified through scRNA-seq data and devised a machine learning-based integration technique to utilize external bulk RNA-seq datasets for the development of hub RLRs. We fitted 150 diagnostic models using 12 distinct machine learning algorithms, employing the GEO series GSE57148 as the training dataset. This process enabled us to screen for hub genes among the 93 RLRs and validate them using the GSE162635, GSE151052, and GSE239897 series. The machine learning results indicated that the RF + plsRglm, RF + glmBoost, and RF + Stepglm[both] models ranked top three based on AUC evaluation. Additionally, we assessed the machine learning models using multiple metrics such as accuracy, recall, precision, and F1 score. The composite model evaluation results showed that RF + glmBoost exhibited the best diagnostic performance, with an AUC of 0.769, accuracy of 0.713, recall of 0.707, precision of 0.799, and an F1 score of 0.745 (Fig. 5A and Additional file 2: Fig. S2). In contrast, the RF + plsRglm (with AUC of 0.796, accuracy of 0.710, recall of 0.673, precision of 0.822, F1 score of 0.732) and RF + Stepglm[both] (with AUC of 0.767, accuracy of 0.691, recall of 0.671, precision of 0.801, F1 score of 0.721) models showed relatively poorer diagnostic stability (Additional file 2: Fig. S2). Following this, we conducted ROC curve analysis on the 16 RLRs identified by the RF + glmBoost algorithm (CBX8, EHD4, HDLBP, KDM6B, LSP1, NAP1L1, NFAT5, NLRP3, NUP214, PAFAH1B3, PINX1, PLD1, POLB, RCC2, RNF213, and VIM) (Additional file 2: Figure S3). Of these, LSP1 (AUC = 0.597, 95%CI: 0.512–0.6716) and NAP1L1 (AUC = 0.557, 95%CI: 0.474–0.641) were excluded due to excessively low ROC scores. Consequently, 14 RLRs were ultimately included (CBX8, EHD4, HDLBP, KDM6B, NFAT5, NLRP3, NUP214, PAFAH1B3, PINX1, PLD1, POLB, RCC2, RNF213, and VIM), with eight genes upregulated (EHD4, HDLBP, KDM6B, NFAT5, NLRP3, NUP214, PLD1, and RNF213) and six downregulated (CBX8, PAFAH1B3, PINX1, POLB, RCC2, and VIM) in COPD (Fig. 5B).
Fig. 5
Development and evaluation of diagnostic model based on RLRs in COPD. (A) AUC values for 150 combinations of machine learning algorithms across four testing groups. (B) Expression patterns of 14 hub RLRs in the training dataset GSE57148. (C) Nomogram constructed by the 14 hub RLRs. (D-E) DCA analysis to evaluate the accuracy of the diagnostic model. (F-I) ROC curve analysis to assess the diagnostic specificity of the training dataset GSE57148 and validation datasets (GSE162635, GSE151052, GSE239897, GSE76925). Abbreviation: AUC: area under curve; COPD: chronic obstructive pulmonary disease; DCA, decision curve analysis; RLRs: R-loop regulators; CBX8: chromobox 8; EHD4: EH domain containing 4; HDLBP: high density lipoprotein binding protein; KDM6B: lysine demethylase 6B; NFAT5: nuclear factor of activated T cells 5; NLRP3: NLR family pyrin domain containing 3; NUP214: nucleoporin 214; PAFAH1B3: platelet activating factor acetylhydrolase 1b catalytic subunit 3; PINX1: PIN2/TERF1 interacting telomerase inhibitor 1; PLD1: phospholipase D1; POLB: DNA polymerase beta; RCC2: regulator of chromosome condensation 2; RNF213: ring finger protein 213; VIM: vimentin
We further validated the diagnostic model constructed by 14 RLRs and performed potential confounding factor analysis. Firstly, a nomogram was constructed based on the expression of these 14 RLRs. Among them, the value of each characteristic variable is associated with a score point, and the total score is obtained by adding up the scores of all characteristic variables, representing the risk of COPD (Fig. 5C). DCA confirmed that the clinical application of the nomogram brought certain clinical benefits to COPD patients (Fig. 5D-E). We also integrated the prediction model score and important clinical characteristics to construct a prediction nomogram to evaluate potential confounding factors. The results showed that the prediction model is an independent predictor for the diagnosis of COPD. Secondly, the diagnostic specificity and sensitivity of the diagnostic model itself were assessed using ROC curves. The evaluation revealed that the ROC of the diagnostic model in the training set is 0.904 (Fig. 5F); the results of the three test sets show that the ROC of GSE162635 is 0.802 (Fig. 5G), the ROC of GSE151052 is 0.628 (Fig. 5H), the ROC of GSE239897 is 0.644, the ROC of GSE76925 is 0.635 (Fig. 5I). Finally, we calculated the AUC and its corresponding 95% CI for each gene in the training set (Additional file 2: Figure S3), as follows: CBX8 (AUC = 0.731, 95% CI: 0.659–0.793), EHD4 (AUC = 0.779, 95% CI: 0.714–0.842), HDLBP (AUC = 0.770, 95% CI: 0.698–0.834), KDM6B (AUC = 0.674, 95% CI: 0.595- 0.793), NFAT5 (AUC = 0.682, 95% CI: 0.605–0.756), NLRP3 (AUC = 0.690, 95% CI: 0.614–0.765), NUP214 (AUC = 0.798, 95% CI: 0.730–0.860), PAFAH1B3 (AUC = 0.784, 95% CI: 0.784, 95% CI: 0.698–0.834), KDM6B (AUC = 0.674, 95% CI: 0.595–0.834), KDM6B (AUC = 0.674, 95% CI: 0.595- 95% CI: 0.716–0.845), PINX1 (AUC = 0.677, 95% CI: 0.600–0.756), PLD1 (AUC = 0.693, 95% CI: 0.617–0.762), POLB (AUC = 0.665, 95% CI: 0.583–0.743), RCC2 (AUC = 0.674, 95% CI: 0.592–0.751), RNF213 (AUC = 0.806, 95% CI: 0.738–0.868), VIM (AUC = 0.657, 95% CI: 0.578–0.737).
Immune Infiltration among Patients with Different R-loop RisksHaving demonstrated the robustness of the R-Loop score model derived from 14 hub RLRs, we subsequently stratified COPD patients into high-risk (low R-loop score) and low-risk (high R-loop score) groups based on the median value of the riskScore. GSEA was applied to identify enriched signaling pathways between these two groups. The riskScore was positively correlated with signaling pathways related to Graft-Versus-Host Disease, Antigen Processing and Presentation, Allograft Rejection, Systemic Lupus Erythematosus, and Leishmaniasis (Fig. 6A), while it was negatively correlated with pathways associated with the Metabolism of Xenobiotics by Cytochrome P450, Chemical Carcinogenesis DNA Adducts, Ribosome, Oxidative Phosphorylation, and Drug Metabolism Other Enzymes (Fig. 6B). Consistently, the immune score of the high-risk COPD group (low R-loop score) was higher than that of the low-risk group (high R-loop score) (Fig. 6C), indicating that individuals at higher risk exhibit a more robust immune response and may benefit from immunotherapy. The infiltration of 22 immune cells in the two groups was analyzed using the CIBERSORT algorithm to study changes in the immune landscape (Fig. 6D).
Fig. 6
Immune infiltration analysis in high-risk and low-risk COPD groups. (A) Top five upregulated pathways by GSEA between high-risk and low-risk COPD groups. (B) Top five downregulated pathways by GSEA between high-risk and low-risk COPD groups. (C) Comparison of immune scores between high-risk and low-risk COPD groups. (D) Stacked histogram of the proportion of immune cells between high-risk and low-risk COPD groups. (E) Violin plots showing the comparison of 22 types of immune cells between high-risk and low-risk COPD groups. (F) Correlation between 14 hub genes and 22 immune cells. (G) Heatmap of the correlation between key transcription factors involved in differential transcriptional activity and 14 hub RLRs. Compared with the low-risk COPD groups, * P < 0.05; ** P < 0.01; *** P < 0.001; ns not significant. Abbreviation: COPD: chronic obstructive pulmonary disease; GSEA: Gene Set Enrichment Analysis; RLRs: R-loop regulators; CBX8: chromobox 8; EHD4: EH domain containing 4; HDLBP: high density lipoprotein binding protein; KDM6B: lysine demethylase 6B; NFAT5: nuclear factor of activated T cells 5; NLRP3: NLR family pyrin domain containing 3; NUP214: nucleoporin 214; PAFAH1B3: platelet activating factor acetylhydrolase 1b catalytic subunit 3; PINX1: PIN2/TERF1 interacting telomerase inhibitor 1; PLD1: phospholipase D1; POLB: DNA polymerase beta; RCC2: regulator of chromosome condensation 2; RNF213: ring finger protein 213; VIM: vimentin
Between the two groups, significant differences were observed in the infiltration of activated DCs and neutrophils in lung tissue (P < 0.05) (Fig. 6E), with activated DCs significantly enriched in the microenvironment of high-risk (low R-loop score) COPD group, and neutrophils significantly enriched in the microenvironment of low-risk (high R-loop score) COPD group. We further evaluated the potential relationships between the 14 hub RLRs and immune cells (Fig. 6F). Correlation analysis based on the CIBERSORT algorithm scores for immune cells and biomarkers showed that activated DCs were positively correlated with KDM6B and negatively correlated with HDLBP (P < 0.01). Neutrophils were positively correlated with VIM, RNF213, NUP214, NLRP3, KDM6B, HDLBP, and EHD4, and negatively correlated with RCC2, PAFAH1B3, and CBX8 (P < 0.01). Further analysis in GSE57148 revealed correlations between these TFs and the 14 hub RLRs. ARID3A, BHLHE41, CREM, EGR1, JUNB, MAFB, MAFF, MYBL2, NFKB1, TFEC and XBP were found to be positively correlated with most of the 14 RLRs, suggesting the existence of a potential positive regulatory network. Conversely, E2F1, JUND, KLF5, and RFX2 were negatively correlated with the majority of the 14 hub RLRs (Fig. 6G).
Identification of Specific Chemotherapeutic Agents Associated with R-loops in COPDReduced R-loop scores are associated with increased risk of COPD, underscoring the urgent need for therapeutic strategies targeting R-loop related mechanisms. To identify small molecule drugs that could reverse the expression pattern of RLRs, we utilized the CMAP database to explore potential therapeutic agents for COPD patients in both high-risk (low R-loop score) and low-risk (high R-loop score) groups. For the high-risk group (low R-loop score), the top 10 drugs with personalized treatment potential include 1-phenylbiguanide, AS-703026, BAS-09104376, CAY-10618, clobenpropit, Memantine, PD-032590, PD-184352, QL-XI-92 and U-0124 (Fig. 7A), primarily targeting pathways such as MEK inhibitor, Histamine receptor antagonist, Serotonin receptor agonist, DDR1 inhibitor, Glutamate receptor antagonist, HIV integrase inhibitor and NAMPT inhibitor (Fig. 7B). The specific molecular structures of these ten molecules are depicted in Fig. 7C-L. For the low-risk group (high R-loop score), the most effective therapeutic drugs include AG-879, anisomycin, helveticoside, iloperidone, MLN-4924, periplocymarin, phorbol-12-myristate-13-acetate, QL-X-138, radicicol and ruxolitinib, mainly involving target pathways such as mTOR inhibitor, DNA synthesis inhibitor, NEDD activating enzyme inhibitor, ATPase inhibitor, PKC activator, Apoptosis stimulant, JAK inhibitor, HSP inhibitor, Angiogenesis inhibitor, and Dopamine receptor antagonist (Additional file 2: Figure S4). Supplementary Fig. 4 illustrate the specific molecular structures of these ten molecules. Specifically, PD-0325901 and QL-X-138 exhibit the lowest CMAPscores in the high-risk (low R-loop score) and low-risk (high R-loop score) groups, respectively, demonstrating the maximal therapeutic benefit for COPD patients with differing R-loop scores.
Fig. 7
Identification of potential small molecule compounds for high-risk COPD (low R-loop score) group through CMAP analysis. (A) The top 10 compounds with the highest negative enrichment scores. (B) Descriptions of pathways associated with the 10 compounds. (C) Chemical structures of the 10 compounds. Abbreviation: CMAP: connectivity map; COPD: chronic obstructive pulmonary disease
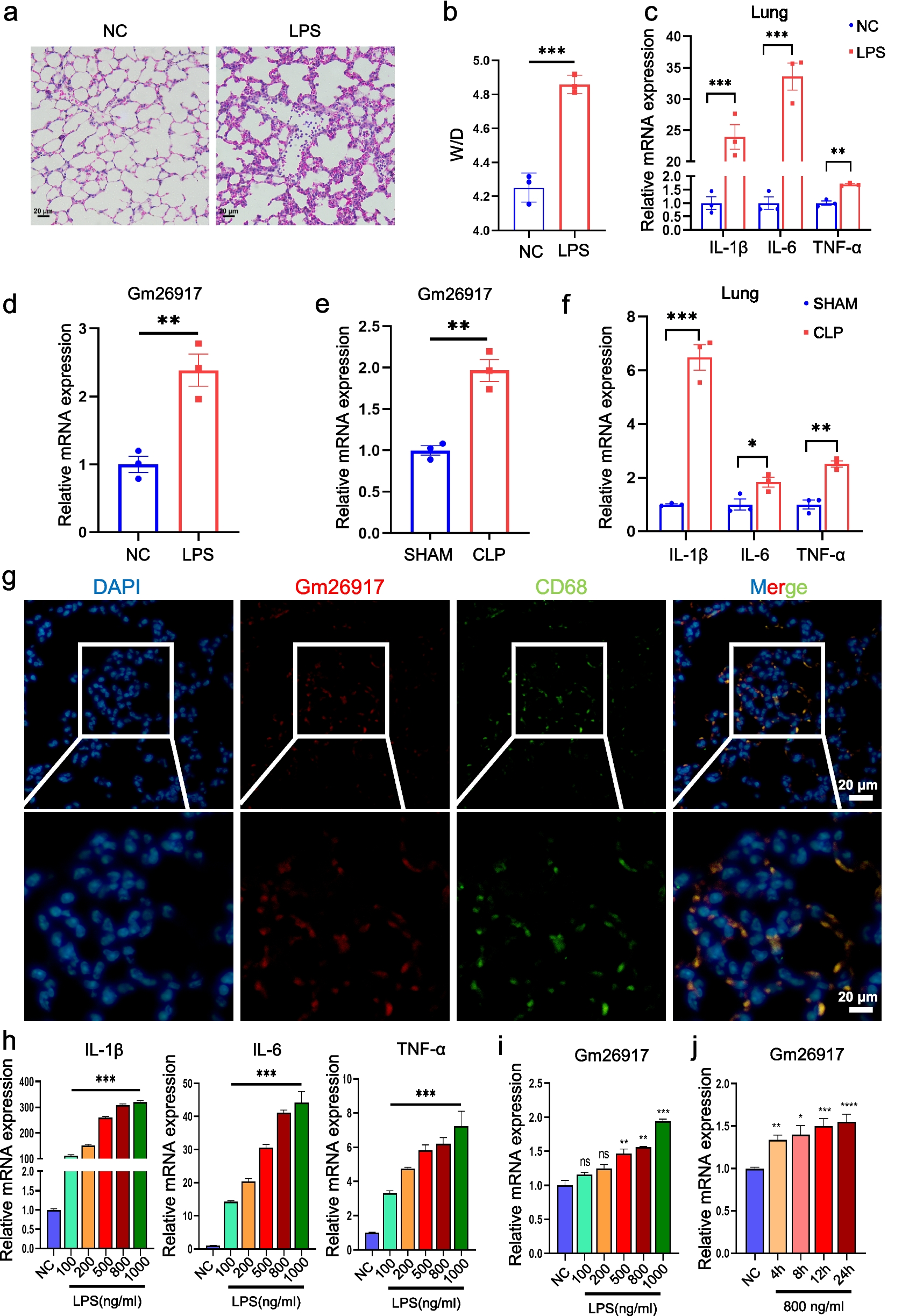
Validation of 14 RLRs by RT-PCRFinally, we further validated the levels of R-loops and the expression of these 14 hub RLRs in the lung tissues of COPD mice. The animal experimental design of the study is shown in Fig. 8A, with representative images of hematoxylin–eosin staining of lung tissue presented in Fig. 8B. Compared to the control group, COPD mice exhibited a significant increase in mean linear interval and an elevated lung pathology score (Fig. 8B), indicating the successful establishment of the COPD mice model. Immunofluorescence results suggest that, compared to control mice, the levels of R-loops in the lung tissues of COPD mice are significantly elevated (Fig. 8C-D). RT-PCR results further demonstrated that, compared to the control group, the mRNA expression of EHD4, HDLBP, NFAT5, NLRP3 and PLD1 was significantly up-regulated, while the mRNA expression of PINX1, POLB and VIM was significantly down-regulated in the lung tissue of COPD mice (all P < 0.05) (Fig. 9A-N).
Fig. 8
Increased R-loop formation in the lungs of mice. (A) Schematic representation of Control and COPD mice. (B) Representative images of hematoxylin–eosin staining sections in lung tissues of Control and COPD mice, along with analysis of Mean liner intercept analysis and Lung pathological scores (n = 3 per group). (C) Immunostaining of R-loops in lung tissue sections from control and COPD mice using S9.6 antibody and quantification of fluorescence intensity (n = 3 per group). Data are expressed as mean ± SD, Student's t-test. Compared with the control group, * P < 0.05; ** P < 0.01; *** P < 0.001. Abbreviation: COPD: chronic obstructive pulmonary disease; LPS: lipopolysaccharide; SD: standard deviation
Fig. 9
Expression of 14 hub RLRs in lung tissue of COPD mice. (A-N) CBX8 (A) EHD4 (B) HDLBP (C) KDM6B (D) NFAT5 (E) NLRP3 (F) NUP214 (G) PAFAH1B3 (H) PINX1 (I) PLD1 (J) POLB (K) RCC2 (L) RNF213 (M) VIM (N) (n = 3 per group). Data are expressed as mean ± SD, Student's t-test. Compared with the control group, * P < 0.05; ** P < 0.01; **** P < 0.0001; ns: not significant. Abbreviation: COPD: chronic obstructive pulmonary disease; LPS: lipopolysaccharide; SD: standard deviation; CBX8: chromobox 8; EHD4: EH domain containing 4; HDLBP: high density lipoprotein binding protein; KDM6B: lysine demethylase 6B; NFAT5: nuclear factor of activated T cells 5; NLRP3: NLR family pyrin domain containing 3; NUP214: nucleoporin 214; PAFAH1B3: platelet activating factor acetylhydrolase 1b catalytic subunit 3; PINX1: PIN2/TERF1 interacting telomerase inhibitor 1; PLD1: phospholipase D1; POLB: DNA polymerase beta; RCC2: regulator of chromosome condensation 2; RNF213: ring finger protein 213; VIM: vimentin
留言 (0)