
記住我

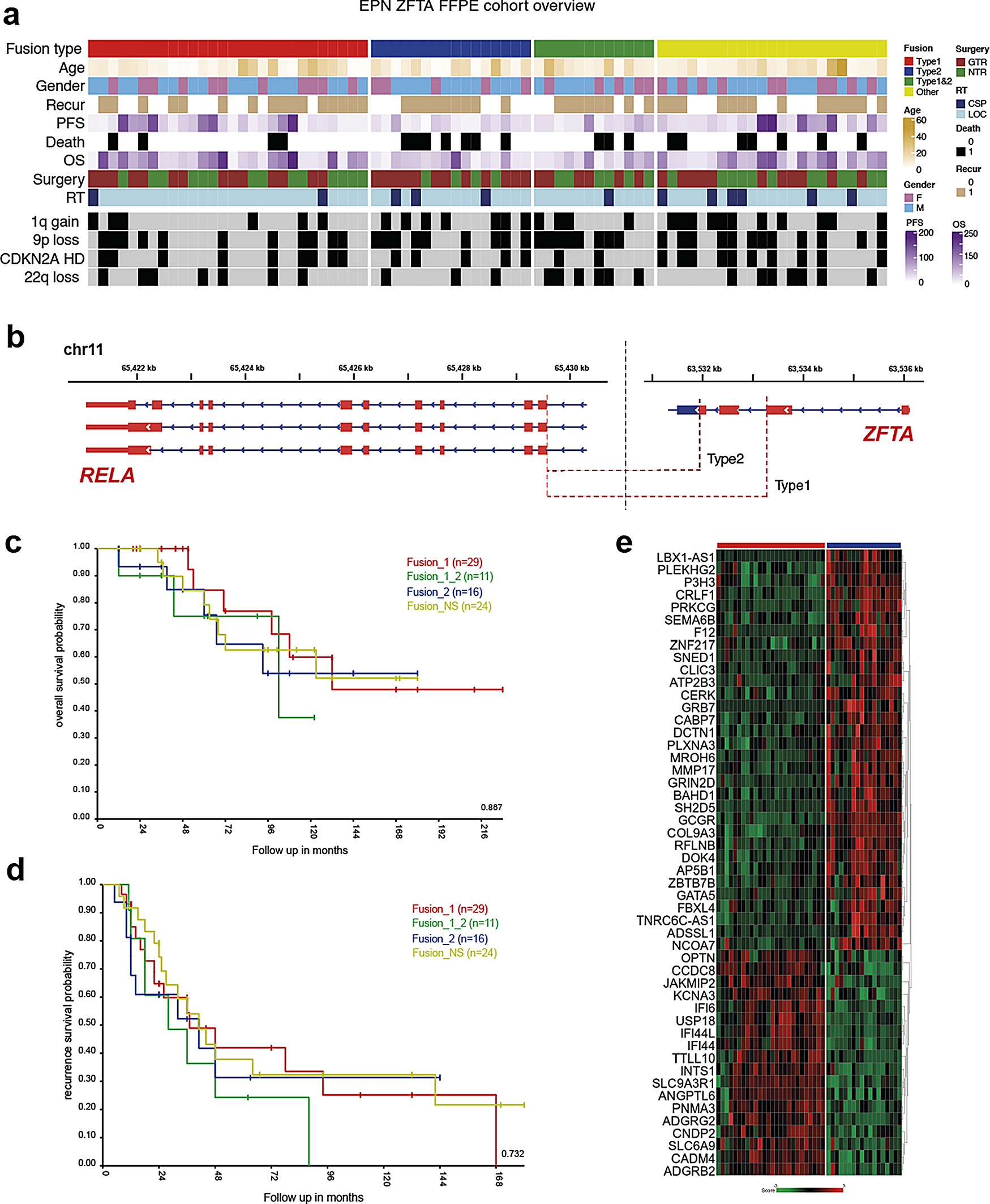




The clinical and molecular characteristics of 80 patients with ST-EPN ZFTA-RELA are summarized in Fig. 1a; Table 1 and Suppl. Table 1. Patients were aged between 4 and 64 years (median: 11.3), with a preponderance of patients younger than 18 years (85% vs. 15% adult patients), and a male: female ratio of 2.5:1. Only a minority of patients (4%) were diagnosed as M2-3 stages at initial presentation. All 80 patients were treated with maximal safe surgical resection and received postoperative radiotherapy (RT), either conformal local RT in 68 patients (85%) or craniospinal RT in 12 patients (15%). Fifty-eight patients (65%) received chemotherapy (HIT-based protocol) after RT. Tumor histology was identified as anaplastic EPN (EPN Grade 3). Dot-like EMA, membranous L1CAM, and nuclear p65-RelA expression were identified in all samples analyzed. Disease relapses occurred in 47 of the patients (60%) and 41 relapsed patients (88%) were treated with second-line surgery, re-irradiation (either conformal or radiosurgery), and/or chemotherapy with various regimens. Twenty-two (28%) of relapsed patients succumbed to disease, 33 patients (41%) showed “no evidence of disease” at last follow-up, and 25 patients (31%) were “alive with disease”. Recurrent copy number variants (CNVs) observed in > 20% of cases were 1q gain (35%), 9p loss (50%) accompanied with 9p21/CDKN2A/B homozygous deletion in 30%, 22q loss (30%), and monosomy X (30%). In line with previous retrospective studies [27, 30], 5-year PFS was 45%, 5-year OS was 82%, and 10-year OS was 61% for this cohort, and no clinical or cytogenetic variables were associated with patients’ survival (Table 2).
Table 1 Clinical and molecular variables for ST-EPN ZFTA-RELA with various fusion variantsTable 2 Uni- and multivariate overall survival analyses for ZFTA-RELA-fused ST-EPN cohortTypes of ZFTA-RELA fusions in ST-EPN and their clinical-molecular characteristicsBy RNA sequencing, several distinct variants of the ZFTA-RELA fusions were identified (Fig. 1b): (i) fusion type 1 – ZFTA exons 1_2 and RELA exons 2_11 (29/35%); (ii) fusion type 2 – ZFTA exons 1_3 and RELA exons 2_11 (16/20%); (iii) combined type_1 and type_2 fusions (11/15%), designated as fusion type 3; (iv) other less common ZFTA-RELA fusion variants were designated as fusion type 4 (24/30%) (Table 1). Some fusions with involvement of ZFTA and other genes were identified in an additional set of ST-EPN cases (n = 14) reflecting previously reported results [37], but this subset was excluded from further analysis (data not shown). Among CNVs, 1q gains and losses of 9p were frequent in ST-EPN ZFTA-RELA with fusion type 2 (55% and 60%, respectively). Treatment details were similar for all ST-EPN ZFTA-RELA fusion types and no survival differences were identified between the various fusion variants (Fig. 1c, d).
Fig. 1
a) Annotation onco-plot describing patient histological and molecular characteristics for target ZFTA-RELA ST-EPN tumors with available RNA sequencing data (n = 80). The following abbreviations were used: RT - radiotherapy, LOC - conformal local, CSP - craniospinal, PFS—progression-free survival, CNV—copy number variants. b) Genomic locations the ZFTA-RELA fusion breakpoints stating the main types of the fusion. c, d) No survival differences were identified between the various ZFTA-RELA fusion types. d) Heatmap of significant differentially expressed genes between ZFTA-RELA fusion type 1 (n = 29) and 2 (n = 16)
Genes differentially expressed between various fusion types of ST-EPN ZFTA-RELAComparing transcriptome profiles generated for ST-EPN with ZFTA-RELA fusion types 1 (n = 29) and 2 (n = 16), 134 genes and processed pseudogenes were identified as differentially expressed genes (DEG) between these molecular variants; 98 were overexpressed in ST-EPN ZFTA-RELA with fusion type 1, and 36 in ST-EPN ZFTA-RELA with fusion type 2 (Fig. 1e; Suppl. Table 2). Thus, INTS1, CCDC8, ADGRG2, KCNA3 were top-ranked genes in ST-EPN ZFTA-RELA type 1, whereas CRLF1, GCGR, PRKCG, GRIN2D were the top overexpressed genes for ST-EPN ZFTA-RELA type 2. In turn, transcriptome signatures of ST-EPN ZFTA-RELA type 1 identified with Gene ontology analysis included pathways involved in the cilium/axoneme, immune response, interferon synthesis, response to viral stimulus, and RNA binding. In contrast, signaling pathways identified for ST-EPN ZFTA-RELA type 2 were enriched with genes involved in neuron guidance, tyrosine kinase, transmembrane transport, and phosphorylation (Suppl. Table 3). There were no statistically significant differentially expressed genes identified between ST-EPN ZFTA-RELA with other fusion types, perhaps due to significant molecular variability within ST-EPN fusion groups 3 and 4.
Gene sets associated with survival ST-EPN ZFTA-RELAMultiple gene OS analysis (see Methods) identified 1892 survival-associated genes with BGN on the top of the list (Suppl. Table 4). Among them, 253 genes disclosed independent hazard ratios (HR) by Cox regression analysis. In total, 1545 genes (147 with independent HR) were associated with favorable OS; among them, genes of the coiled-coil domain containing family (CCDC; n = 16), family with sequence similarity (FAM; n = 25), keratin family (KRT; n = 21), small nucleolar RNA family (SNORD; n = 15), and zinc finger protein family (ZNF; n = 23) prevailed. In contrast, 347 genes (106 with independent HR) were defined as unfavorable molecular indicators; among them, mitogen-activated protein kinase family (MAPK; n = 11), protocadherin family genes (PCDH; n = 11), and ribosomal protein family L/S (RPL/RPS; n = 11) were frequent. Moreover, 1423/1892 (75%) of these genes were also associated with PFS in ST-EPN ZFTA-RELA. Supervised k-mean clustering defined a set of 100 genes (metagene set) which subdivided ST-EPN ZFTA-RELA into two transcriptome subtypes with favorable/standard (n = 52; 5-year PFS – 50%; 5-year OS – 100%; 10-year OS – 75%) and unfavorable (n = 28; 5-year PFS – 10%; 5-year OS – 30%; 10-year OS − 0) clinical outcomes (Fig. 2a-c; Table 2). The favorable subtype was associated with fusion type 1 (45%), whereas the unfavorable subtype disclosed frequent 1q gain (60%), and fusion type 2 (45%) (Table 3). There were no associations between the prognostically relevant transcriptome subtypes and other clinical-molecular variables. DEG analysis identified 232 genes with BGN as the top-ranked gene within the unfavorable subtype and INTU – within the favorable subset (Fig. 2d; Suppl. Table 5). By gene ontology analysis, the favorable ST-EPN subtype was associated with cilium motility and assembly, axoneme, and cytoskeleton microtubule pathways, whereas the unfavorable – with the extracellular matrix, collagen metabolism, angiogenesis, and cell migration/motility, pathways (Suppl. Figure 1a; Suppl. Tables 6 and 7). Cell type-specific gene set expression analysis (GSEA) disclosed that the favorable subtype was enriched with transcriptome signatures of ciliated epithelial and neuroepithelial cells, human radial glial cells, and cortex embryonic astrocytes, whereas the unfavorable subtype was enriched in signatures of mesenchymal stromal cells, embryonic brain endothelial and microglial cells, and embryonic neural stem cells (Suppl. Table 8). By inspection of methylation level between favorable and unfavorable cases it was possible to identify n = 656 differential CpG sites (Suppl. Table 9), however overlap with detected DEGs locations was only 2%.
Fig. 2
Supervised k-mean clustering of multigene survival data (a) defined a set of 100 genes (metagene set) that subdivided ST-EPN RELA into two transcriptome subtypes (TRS): favorable (n = 52) and unfavorable (n = 28). Two identified TRS were associated with patients’ OS (b) and PFS (c). d) Heatmap of top 20 most confident genes differentially expressed between clinically relevant TRS with BGN on the top of this list
Table 3 Clinical and molecular variables for ST-EPN ZFTA-RELA relevant transcriptome subtypesCell content differences in clinically relevant transcriptome ST-EPN ZFTA-RELA subtypesWe performed deconvolution analysis of bulk RNA-seq data to decipher ST-EPN ZFTA-RELA inter- and intra-tumoral cellular heterogeneity. For this purpose, we used a published single-cell RNA-seq dataset [14] that was composed of ST-EPN ZFTA-RELA (n = 5) covering 10 clusters of neoplastic cells that exhibited molecular signatures matching different transcriptome metaprograms (see below). Deconvolution analysis of bulk RNA profiles was performed using the BayesPrism computational program (see Methods), and significant proportions of neoplastic cells (more than 80%) were identified in all tumor samples. The proportion of non-tumoral cells was quite low (median ~ 7%). Based on the deconvolution analysis of single-cell molecular signatures, the bulk RNA-seq ST-EPN dataset was composed of two mitotic/proliferative cell programs (ST-S-Phase – 5% and ST-G2/M-Phase – 5%), two progenitor cell programs (ST-Radial-Glia-Like – 15% and ST-Neuronal-Precursor-Like – 15%), differentiated cell programs (ST-Ependymal-Like – 10%), interferon signaling program (ST-Interferon-Response – 10%), metabolic program (ST-Metabolic – 10%), and extracellular matrix program (ST-RELA-Variable – 20%) (Fig. 3a). A higher than median proportion of ST-RELA-Variable cell type subpopulation conferred the shortest OS (p < 0.01; Suppl. Figure 1b), whereas high ST-Ependymal-Like and ST-Interferon-Response cell fractions were associated with favorable clinical outcomes (p < 0.01 and p < 0.01 respectively; Suppl. Figure 1c, d). In addition, the shortest PFS but not OS was identified for higher ST-Radial-Glia-Like subpopulation (p < 0.01; not shown).
We further analyzed cell content within clinically relevant ST-EPN transcriptome subtypes (Fig. 3a-e; Table 3). In this analysis, the unfavorable subtype showed higher proportion of ST-RELA-Variable (35% vs. 15%; p < 0.01) cell subpopulation (Fig. 3b). In contrast, the clinically favorable ST-EPN subtype was composed of differentiated ST-Interferon-Response (12% vs. 3%; p < 0.01), ST-Radial-Glia-Like (27% vs. 12%; p < 0.01) and ST-RELA-Ependymal-like (10% vs. 5%; p < 0.01) cell subpopulations (Fig. 3c-e). There were no differences in other cell subpopulations between clinically relevant ST-EPN subtypes.
Fig. 3
a) Bar plots of predicted relative proportions of EPN ZFTA cell types in bulk tumor gene expression profiles. Annotation provides favorable (FAV; blue) and unfavorable (UFV; red) status for each tumor sample. (b-e) Boxplots of statistically significant differences between EPN ZFTA favorable and unfavorable in proportions of ST-RELA-Variable (b), ST-Interferon-Response (c), ST-Radial-Glial (d) and ST-Ependymal (e) neoplastic cell subpopulations
To verify the deconvolution results detected for the ST-EPN cohort, gene set variance analysis (GSVA) was performed as an alternative computational method on mean gene expression values computed from RPKM matrices generated for favorable and unfavorable transcriptome ST-EPN subsets, as described (see Methods). GSVA results showed the enrichment patterns in expression signatures of the identified neoplastic cell subpopulations within the clinically relevant transcriptome subtypes reflecting the results of bulk RNA deconvolution analysis for ST-RELA-Variable, ST-Interferon-Response and ST-Radial-Glia-like (Suppl. Figure 1e).
BGN expression as a possible biomarker for ST-EPN ZFTA-RELA risk stratificationBGN was identified by multiple gene survival testing (see Methods) as a top gene associated with poor outcomes (HR 17.85 for PFS and 45.48 for OS; log-rank; p-value < 0.01; see. Suppl. Table 4; Suppl. Figure 2a, b), and was also significantly overexpressed in the unfavorable transcriptome ST-EPN subtype (Suppl. Figure 2c). This gene is known to be associated with maintenance of the extracellular matrix structure and located on chromosome X. Nevertheless, the overexpression of BGN in unfavorable subset was observed as significantly independent of patients’ sex (Suppl. Figure 2d, e). Across ST-EPN cell types BGN expression was found to be active across all tumor cell types, mostly enriched in ST-RELA-Variable and cell cycle-associated subpopulations, but almost not expressed in normal cells fraction (Suppl. Figure 2f). No significant difference in BGN expression was seen between ZFTA-RELA and ZFTA-non RELA ST-EPN (Suppl. Figure 2 g), but gene expression for ST-EPN ZFTA-RELA was significantly higher as compared to ST-EPN-YAP1 and ST-EPN-SE (Suppl. Figure 2 h).
There were no associations between BGN expression levels and DNA profiles at the gene location (Xq28). However, we identified a negative correlation between BGN expression and methylation levels of two CpG sites within the gene promoter region (cg21179255 and cg04177332; Suppl. Figure 3a, b). Moreover, low methylation levels for these two CpGs were associated with poor OS (log-rank; p-value = 0.01 and < 0.01 respectively) (Table 2; Suppl. Figure 3c, d). Nevertheless none of the CpGs lying within BGN loci were significantly differentially methylated between favorable and unfavorable ST EPN transcriptome subtypes (Suppl. Table 9), thus suggesting only an inverse correlation between gene expression levels and methylation of a few GpGs within the promoter region as association.
In a Cox regression model accounting for all clinical and molecular data, the unfavorable ST-EPN subtype and BGN expression were independently associated with poor survival (Table 2). Further, we compared stratification regression models with and without these independent variables. The inclusion of these molecular parameters significantly improved outcome prediction for the current ST-EPN cohort thus reducing prediction errors. Similar results were obtained when we compared receiver areas under curves (AUC) and operating characteristic curves (ROC) for the Cox models at different time points. Thus, the inclusion of the transcriptional subtype and/or BGN expression data resulted in the improvement of the ST-EPN risk stratification model.
In addition, survival analyses of public gene expression data generated with the Affymetrix platform for multi-institutional extended ST-EPN cohort [27] also showed unfavorable outcomes for tumors with high BGN expression, thus confirming data obtained with our RNA sequencing analysis (Suppl. Figure 3e).
IHC with biglycan a possible tool for ST-EPN ZFTA-RELA prognosticationWe applied a biglycan/BGN antibody (see Methods) to stain 70 samples with accessible tumor sections from the current transcriptome analysis cohort (screening set) and 56 samples from an independent molecularly diagnosed ST-EPN ZFTA-RELA cohort applied in previous studies (validation set) [27, 37].
The two following patterns of BGN immunostaining were detected: (i) Expression was found in the tumor vessels (including microvascular proliferates), and patched collections of tumor cells (n = 41 in the screening set and n = 40 in the validation set; Fig. 4a). These samples were considered BGN-negative. (ii) Diffusely and predominantly dot-like immunostaining throughout the entire tumor (n = 29 in the screening set and n = 16 in the validation set; Fig. 4b). These samples were designated as BGN-positive. Two investigators showed perfect interobserver agreement for this categorization (κ = 1), and we did not find differences in terms of staining intensity across both tumor sets. In addition, 6/16 (40%) ST-EPN with ZFTA-non-RELA fusions were BGN-positive, whereas all ST-EPN-YAP1 studied (n = 18) were BGN-negative.
In the screening set, BGN expression data coincided strongly between mRNA and protein levels (correlation coefficient r = 0.857; p < 0.01; Fig. 4c). In addition, 90% ST-EPN with elevated BGN expression (log2 > 4) were BGN-positive in contrast to 5% samples with low gene expression (p < 0.01). Also, most of the ST-EPN (86%) from the unfavorable transcriptome subtype were BGN-positive as compared to 12% ST-EPN allocated to the favorable subtype (p < 0.01).
Survival analysis revealed that BGN-positivity is significantly associated with worse clinical outcomes in both the screening (5-year PFS – 15% and 5-year OS – 45%) and validation (5-year PFS – 10% and 5-year OS – 40%) sets (Fig. 4d-g). Thus, the results of BGN IHC prognostic evaluation correlated closely with the survival data obtained by transcriptome analysis.
Fig. 4
Two variants of BGN protein immunostaining were detected: (a) Negative - expression in the tumor vessels predominantly. (b) Positive - intense dot-like BGN expression in tumor cells. BGN expression levels were significantly higher in immunopositive ST-EPN RELA (c). Survival analysis revealed that BGN immunopositivity is significantly associated with worse clinical outcomes in both the screening (d,e) and validation (f,g) sets of ST-EPN RELA
留言 (0)