
記住我





The p.P36R mutation in ANXA11 is recognized as a hotspot mutation, first reported by Zhang et al. [5] in three patients with ALS or ALS-frontotemporal dementia (ALS-FTD). To date, ten cases of this mutation have been documented [1, 3, 5]. We summarized the demographic characteristics of these cases (Table 1). The mean age of onset for ALS patients carrying this mutation was 72.3 ± 5.5 years, with an average survival time of 29.6 ± 7.9 months. ALS-FTD was observed in six patients (60%), while three patients (30%) presented with cognitive impairment. The site of disease onset was bulbar in seven patients (70%) and limb in three patients (30%). Additional manifestations, such as bradykinesia and hypophonia, have also been reported in a single FTD patient harboring the p.P36R mutation [15]. Patients with mutations in the low-complexity domain (LCD) of ANXA11 adjacent to P36R also present with inclusion body myopathy [2, 16, 17]. A gain-of-function mechanism has been proposed for ANXA11 mutations, as ANXA11 expression remains unaltered alongside the observed pathological findings in patients [1]. Overall, ANXA11-associated ALS is characterized by a late age of onset and extended survival, with multisystem involvement.
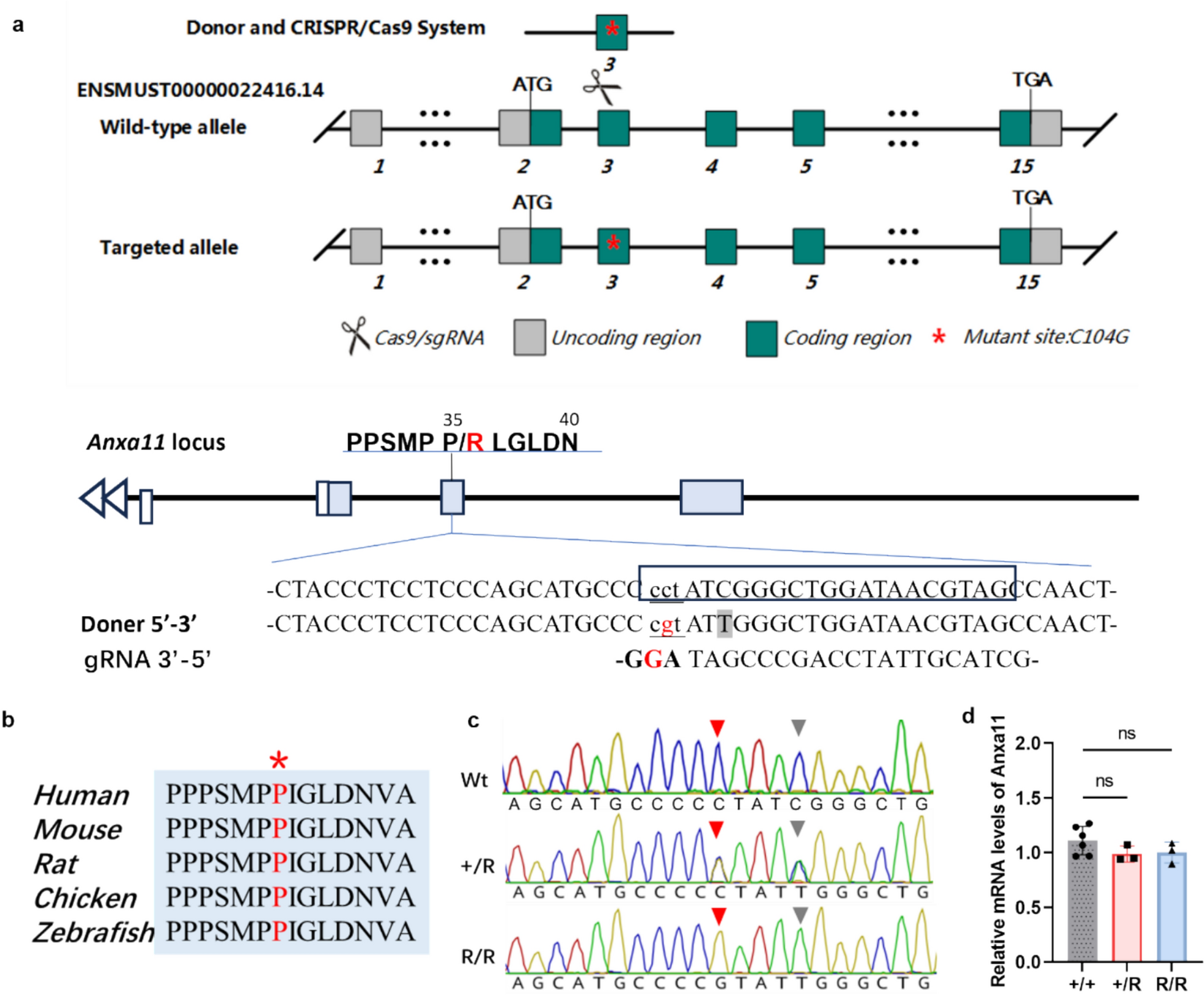
Table 1 Demographic and clinical characteristics of ALS patients with P36R mutation in ANXA11Generation of a knock-in mouse line carrying the ANXA11-P36R mutationThe P36R mutation in ANXA11 is located in the LCD and the amino acid is conserved across species (Fig. 1A, 1B). The mouse position corresponding to human P36 is P35. The position is a PAM site for Cas9 recognition. The correct sequence (P35 to R) was introduced and germ line transmission was confirmed after genome editing (Fig. 1C). The Anxa11 mRNA expression level was comparable with that of wt either in heterozygous (het, + /R) or homozygous (hom, R/R) mutant mice in cortex (Fig. 1D). The protein level of mouse ANXA11 was not affected compared with that of the wt in cortex and muscle of mutant mice in both het and hom mice (Supplemented Fig. 1). Also, homologous PCR products with primers located in exon 1 and exon 4 of Anxa11 showed that no splicing mutation was introduced. We therefore conclude that a knock-in mouse line carrying the ALS ANXA11-P36R mutation is established.
ANXA11-P36R knock-in mice exhibit late-onset motor neuron diseaseSimilar to the late-onset motor disability and extended life expectancy observed in ALS patients carrying ANXA11 mutations [14] (Table 1), ANXA11-P36R knock-in mice demonstrated significant motor decline at 10 months (mo) of age in both het and hom animals (Fig. 2; Supplementary Fig. 2) compared to wt mice, with no evident impairment in younger mice (6 mo; Supplementary Fig. 3). Paralysis was not observed up to 12 mo of age, although occasional hindlimb spasms were noted. Mutant mice displayed reduced limb stretching when suspended by the tail compared with wt mice. Behavioral tests revealed reductions in grip strength and spontaneous locomotor activity in het mice compared with wt (Fig. 2a, b). Hanging endurance time and rotarod performance further indicated diminished motor ability (Fig. 2a). No differences were observed in disease onset or progression speed between het and hom mice, although spasms were more frequent in hom mice from 8–9 mo onward (Supplementary Fig. 4). The knock-in mice also exhibited prolonged survival, with no disease-related mortality observed during a 24-mo observation period; one mouse died due to injury, and one from unknown causes shortly after birth. Overall, ANXA11-P36R mutant mice exhibited relatively mild disease progression without severe paralysis.
Fig. 2
Motor and cognitive performance of 10-month-old homozygous ANXA11-P36R knock-in mice. a Motor decline in homozygous (R/R) mutant mice compared with wild-type (+ / +). Mutant mice showed reduced grip strength (p = 0.0466), shorter hanging endurance time (p = 0.0078), and decreased retention time on the rotarod test (p = 0.0329) compared with wild-type. b Representative heatmap and results from the open field test with no significant difference between the two groups. c The results from the elevated plus maze. Homozygous ANXA11-P36R knock-in mice displayed tendency of anxiety-like behavior with no significance compared with wild-type (p = 0.052). d Dementia was not evident in homozygous 10-month-old mutant mice. The percentage of time spent investigating a novel object (left, NOR), and the number of entries into the novel arm and spontaneous alternation (right, Y maze) showed no significant differences between wild-type and mutant mice. Data are presented as mean ± SEM (n = 8 mice per group), with statistical analysis performed using Student’s unpaired t-test
Electromyography reveals early neurogenic impairment in ANXA11-P36R knock-in mice. Electromyographic assessments were performed on hom ANXA11-P36R knock-in mice and their wt littermates at 2, 4, 10, and 12 months of age. Denervative potentials were first detected in multiple skeletal muscles at 2 months, although CMAP amplitudes remained normal at this stage. By 4 months, mutant mice exhibited abundant fibrillations, positive sharp waves, and spontaneous and giant motor unit potentials across muscles in multiple spinal cord segments, which were scarcely observed in wt littermates (Fig. 3a). Both mutant and wt mice showed motor development from 4 to 10 months, as indicated by increased CMAP. However, mutant mice exhibited a significant decline in baseline-to-peak (B-P) and peak-to-peak (P-P) CMAP amplitudes, with a more pronounced decrease in hindlimbs compared with forelimbs. Notably, a significant reduction in CMAP was first observed in hindlimbs at around 4 months, and in forelimbs at around 10 months (Fig. 3b). These findings suggest early and extensive neurogenic impairment, characteristic of ALS in patients.
Fig. 3
Electromyography of homozygous ANXA11-P36R knock-in mice. A Representative images of denervative potentials detected in mutant mice, including fibrillation potentials in the left shoulder-deltoid muscle (SDM), positive sharp waves (PSWs) in the left erector spinae muscle (ESM) and right gastrocnemius muscle (GAS), and spontaneous and giant motor unit potentials in the right GAS and right rectus femoris muscle (RFM). B Representative CMAP responses in the forelimbs and hindlimbs of wild-type (+ / +) and mutant (R/R) mice. Left, representative CMAP images in wild-type and mutant mice at different ages. Right, peak-to-peak CMAP amplitudes in wild-type and mutant mice. Both mutant and wild-type mice exhibited motor development from 4 to 10 months. CMAP amplitudes decreased significantly in mutant mice over time, particularly in hindlimbs from 10 to 12 months, whereas amplitudes remained stable in wild-type mice (Two-way ANOVA with Sidak’s multiple comparisons test, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; n = 3 mice per group)
Intact working memory was observed in het and hom ANXA11-P36R knock-in mice. To assess FTD-like symptoms, we conducted NOR, Y-maze, and EPM tests. During NOR tasks, both het and hom mice explored objects equally with wt, indicating no difference in novelty discrimination (Fig. 2d, Supplemented Fig. 2d). In the Y-maze test, there was no significant difference in the number of entries into the novel arm or in spontaneous alternations between het and wt, as well as between hom and wt mice (Fig. 2d, Supplemented Fig. 2d). However, at 10 months, het knock-in mice displayed anxiety-like behavior, with significantly fewer entries into the open arms during the EPM test compared with wildtype mice (p < 0.01; Fig. 2c). Hom mice showed a decreasing trend with no statistically significant difference (p = 0.052) compared with the wt in the open arm entry frequency (Supplementary Fig. 2). These findings indicate that ANXA11-mutant mice do not exhibit dementia but display a propensity for anxiety-like behavior.
We thus conclude the established ANXA11-P36R knock-in mice recapitulates key phenotypes of ALS patients, including late-onset motor decline and preserved lifespan.
Early aberrant protein aggregation in the central nervous system of ANXA11-P36R miceIn ALS patients with ANXA11 mutations, ANXA11 forms diverse and heterogeneous aggregates, which are believed to contribute to neurodegeneration [18]. In the ANXA11-P36R knock-in mouse model, we observed early aggregates of mutant ANXA11 in the cytoplasm of spinal cord motor neurons (MNs) at 2 months of age (Fig. 4a). These aggregates co-localized with SQSTM1/p62-positive inclusions (Fig. 4a), which are associated with oxidative stress, the ubiquitin–proteasome system, and the autophagy pathway [19, 20]. In wt littermates, ANXA11 was homogeneously dispersed in the nucleus and cytoplasm, and SQSTM1/p62 protein did not form aggregates (Fig. 4a; Supplementary Fig. 5). The co-localization of mutant ANXA11 aggregates with SQSTM1/p62 inclusions became increasingly prominent in spinal cord MNs as the disease progressed (Fig. 4a). Also, in 4-month-old hom mice, mutant ANXA11 aggregates co-localized with SQSTM1/p62 inclusions in the cortical neuron (Supplementary Fig. 5).
Fig. 4
Early abnormal protein aggregation in central nervous system. a Representative fluorescence images show ANXA11 (green) and p62 (red) aggregates in wild-type (+ / +) and mutant (R/R) mice at various ages in spinal cord motor neurons. In mutant mice, ANXA11 aggregates are evident in the cytoplasm, co-localizing with SQSTM1/p62-positive clumps from 2 months of age, with increased prominence as the disease progresses (right). Line-scan plots depict the extent of ANXA11 and p62 co-localization. b Representative fluorescence images of ANXA11 (green) and TDP-43 (red) aggregates in wild-type (+ / +) and mutant (R/R) mice at different ages. TDP-43 mislocalization from the nucleus to the cytoplasm, partially co-localizing with ANXA11 aggregates, is observed in an age-dependent manner in mutant mice. Line-scan plots (right) illustrate a significant increase in ANXA11 and TDP-43 co-localization in mutant mice from 4 months onwards. c DAB (3,3'-diaminobenzidine) staining showed translocation of TDP-43 from nucleus to cytoplasm from 4 months on in the brain of homozygous (R/R) mutant mice. d Western blotting of nuclear and cytosolic fractions from brains demonstrated an increase in cytosolic TDP-43 levels in homozygous (R/R) mutant mice compared to wild-type (+ / +). GAPDH was used as the marker for the cytosolic fraction, and PCNA for the nuclear. Hoechst was used to label nuclei. Scale bars, 10 μm. N = 3 mice, n = 7 sections per group. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
TDP-43 proteinopathy and ANXA11-TDP43 interaction in ANXA11-P36R knock-in miceMislocalization of TDP-43 is a hallmark of ALS pathology, typically remaining nuclear under normal conditions but translocating to the cytoplasm in neurodegenerative diseases [21]. Unlike SOD1-related ALS, which does not always exhibit TDP-43 proteinopathy [22], ANXA11-P36R knock-in mice displayed TDP-43 mislocalization in the anterior horn of the mouse spinal cord and cortical neurons (Fig. 4b, c, supplemented Fig. 5). We also extracted proteins from the cytoplasm and the nucleus, respectively, for verification. The evaluation of the cellular localization of TDP-43 showed that the level of cytoplasmic TDP-43 in ANXA11-mutant mice increased significantly, and the level within the nucleus decreased remarkably (Fig. 4d).
Fig. 5
Muscle pathology findings in homozygous ANXA11-mutant mice. a Hematoxylin and eosin (HE) staining of gastrocnemius muscle in wild-type and mutant mice. Muscle fibers in 2-month-old wild-type (+ / +) and mutant (R/R) mice were intact (left), with inclusion body myopathy becoming evident from 4 months onward (middle), as indicated by occasional dense sarcoplasmic eosinophilic aggregates (black arrow). By 9 months, mutant mice showed marked muscular dystrophy, nuclear centralization, rimmed vacuoles, and eosinophilic inclusions. N = 3 mice. Scale bar, 20 μm. b Immunofluorescence of muscle fibers stained for ANXA11 and TDP-43. In 4-month-old mutant muscle cells, ANXA11 aggregates dispersed in the sarcoplasm and sarcolemma, co-localized with TDP-43 inclusions. Scale bar, 20 μm. c Ultrastructural findings in ANXA11-P36R-associated myopathy. Longitudinal sections of muscle fibers show normal structures in wild-type mice, subsarcolemmal autophagic material in 4-month-old mutants, and Z-disc dissolution with vacuole formation in 9-month-old mutants. Middle panels show subsarcolemmal electrodense structures in small vacuoles (arrow) in 4-month-old homozygous mutants. Bottom panels show autophagic vacuoles with myelin-like debris or remnant organelles in 4- and 9-month-old muscle cells. Scale bars, 1 μm–500 nm (as indicated). Black frames in (a) and white frames in (b) & (c) denote enlarged areas
TDP-43 can be found within axonal transporting granules where annexin A11 tethers these ribonucleoprotein granules to lysosomes [23, 24] and ANXA11 aggregates were shown to comingle with TDP-43 in CNS neurons [25, 26]. In our ANXA11-mutant mice, from 2 months of age, spinal motor neurons exhibited cytoplasmic TDP-43 inclusions around the nucleus, co-aggregating with mutant ANXA11 (Fig. 4b). By 9 months, nearly all TDP-43 had translocated to the cytoplasm, forming co-aggregates with ANXA11, indicating that pathogenic ANXA11 mutations induce TDP-43 proteinopathy, which worsens as the disease progresses (Fig. 4b). To further verify whether TDP-43 and ANXA11 interacted with each other, co-immunoprecipitation was conducted. Pulldown of TDP-43 and its interacting proteins showed detection of ANXA11, which indicates direct interaction between TDP-43 and ANXA11 in the pathological state. (Supplementary Fig. 6).
To evaluate the aggregation status of TDP-43 in the brains of ANXA11-mutant mice, Western blotting was employed to examine protein samples in the TBS-soluble, sarkosyl-soluble, and sarkosyl-insoluble fractions. Monomeric TDP-43 levels were comparable between wild-type and mutant mice in TBS- and sarkosyl-soluble fractions (n = 3 per group). Sarkosyl-insoluble fractions showed significantly elevated monomeric TDP-43 in mutant mice, indicating increased aggregation (Supplementary Fig. 7).
Inclusion body myopathy in ANXA11-P36R knock-in micePatients with ANXA11 mutations present with inclusion body myopathy (IBM) [2], which we recapitulated in our mutant mice model. No muscle dystrophy was evident until 9 months of age in mutant mice (Fig. 5a). Moreover, the gastrocnemius muscle of the ANXA11-mutant mice exhibited grouped muscle atrophy (Supplementary Fig. 8). Nuclear centralization, moth-eaten intermyofibrillar structures, and rimmed vacuoles with eosinophilic inclusions were observed, exhibiting features similar to those in ANXA11-mutant patients [16, 26]. Notably, at 2 months, while muscle cells appeared intact under HE staining, immunofluorescence detected fine particles of mutant ANXA11 in the sarcoplasm and sarcolemma. By 4 months, these particles had progressed to small aggregates co-localizing with TDP-43 inclusions (Fig. 5b). This novel finding suggests that cerebral cortex, spinal cord, and muscles are all affected early in ANXA11-mutant ALS, underscoring the potential diagnostic value of early muscle biopsy.
Electron microscopy revealed myofibrillar disorganization, Z-disc dissolution, and subsarcolemmal autophagic material in mutant mice (Fig. 5c), consistent with patient biopsies [16, 17, 26]. Subsarcolemmal osmophilic structures and vacuoles, resembling myelin-like debris or remnant organelles, were also observed. Lipid and mitochondrial droplets were noted at the vacuole periphery (Fig. 5c). These findings indicate that ANXA11-P36R ALS is a multisystem disorder characterized by TDP-43 proteinopathy.
Late motor neuron degeneration and neuroinflammation in ANXA11-P36R knock-in miceIn addition to impaired motor abilities, ANXA11-P36R knock-in mice exhibit progressive MN degeneration, detectable by 4 months (Fig. 6a). Alongside a reduction in ChAT-positive MNs in the anterior horn of the spinal cord, mutant MNs exhibit larger somas and fewer dendrites (amplified images, Fig. 6a). Significant MN loss was observed by 9 months compared to wild-type siblings (Fig. 6a, ChAT histogram), indicative of neuronal degeneration.
Fig. 6
Neurodegeneration and neuroinflammation in the lumbar spinal cord of homozygous ANXA11-P36R knock-in mice. a Motor neurons (MNs) in the anterior horn of the spinal cord. MN numbers in homozygous mutant mice (R/R) declined compared with wild-type (+ / +) in 4-month and 9-month mice. Mutant MNs exhibited larger somas and fewer dendrites (amplified images, upper right). Histograms show a reduction in ChAT-expressing MNs. Representative images of GFAP-positive astrocytes b and IBA1-positive microglia c in the ventral spinal cord. Reactive astrocyte and microglial populations increased with disease progression in 2-, 4-, and 9-month-old mutant mice compared with wild-type. The loss of MNs and increase in reactive glial cells became significant in 9-month-old mutants. N = 3 mice, n = 12 sections per group. Scale bar, 200 μm. Data represent mean ± SEM; statistical analysis by t-test. *p < 0.05
Neuroinflammation, marked by increased GFAP- and IBA1-positive cells, was detected early in mutant mice (Fig. 6b, 6c). GFAP and IBA1 expression was significantly elevated in homozygous mutants from 4 months, becoming pronounced by 9 months (Fig. 6a, GFAP & IBA1 histograms). During reactive astrogliosis, GFAP is upregulated, mirroring an increase in both astrocyte number, size and processes [27]. Reactive astrocytes exhibited nuclear translocation of NF-κB (Supplementary Fig. 9a), indicating an inflammatory state influences disease pathogenesis in the current model of ALS. Similarly, mutant microglia showed elevated IBA1 levels and an amoeboid morphology (Supplementary Fig. 9b), a characteristic of neuroinflammation [28]. The ANXA11-P36R knock-in ALS model thus displays late MN degeneration and neuroinflammation associated with disease progression.
Mutant ANXA11 is associated with autophagy impairment and activated mTOR signalingGiven ANXA11’s role in RNA granule transport and its interactions with lysosomes, we explored autophagic dysfunction as a potential mechanism underlying the P36R mutation. Autophagy markers were analyzed in the lumbar spinal cord of 2-, 4-, and 9-month-old mice. Autophagy initiation marker Beclin-1(BECN) and autophagosome formation marker LC3 were normal at 2 months but decreased with disease progression (Fig. 7). Conversely, the autophagy degradation marker p62 showed a progressive accumulation. At 9 months, ANXA11-P36R knock-in mice exhibited significantly more neurons and muscle cells with cytosolic p62- and ANXA11-positive inclusions than wild-type siblings (Figs. 4 and 5), demonstrating a loss of autophagic compensation as the disease advanced.
Fig. 7
Progressive autophagy impairment in homozygous ANXA11-P36R knock-in mice. Autophagy markers, including Beclin-1, LC3BII/I, and p62, were analyzed in the cortex of 2-, 4-, and 9-month-old mutant mice, revealing a progressive decline in autophagic activity. Representative immunoblots demonstrate the impairment of autophagy in mutant mice, with quantified levels normalized to GAPDH. Statistical analysis was conducted using one-way ANOVA, with data presented as mean ± SEM (n = 4 biologically independent replicates per group). Significance levels: *p < 0.05, **p < 0.01, ***p < 0.001
MTOR pathway activation in ANXA11-P36R knock-in homozygous mice.Concurrent with autophagy impairment, the Akt/mammalian target of rapamycin (mTOR) signaling pathway, a key modulator of autophagy [29], was increasingly activated in 9-month-old mutant mice. Elevated ratios of phosphorylated mTOR (S2448) to total mTOR were observed, alongside significant increases in mTORC1-dependent phosphorylation of RPS6KB1/S6K (p70 S6K) (Fig. 8), indicative of mTORC1 activation. Additionally, as mTORC1 activation requires recruitment to lysosomal membrane as well as phosphorylation by the upstream kinase AKT [30], we examined the phosphorylation of AKT (p-AKT) at Ser473. The level of p-AKT was significantly elevated (Fig. 8), further supporting Akt/mTORC1 pathway activation. These data suggest that as the disease progresses, the Akt/mTORC1 pathway becomes increasingly activated, paralleling autophagy impairment and motor decline.
Fig. 8
MTOR activation in homozygous ANXA11-P36R knock-in mice. In the cortex of 2-month-, 4 month- and 9-month-old mutant mice, mTOR pathway was activated indicated by elevated p-mTOR/mTOR, p-AKT/AKT, and p-P70S6K/P70S6K levels. Representative immunoblots illustrate these alterations, with data presented as mean ± SEM (n = 4 biologically independent replicates per group). One-way ANOVA was used to determine statistical significance. *p < 0.05, **p < 0.01, ***p < 0.001
In summary, we generated the first ANXA11-P36R knock-in mouse model, providing detailed insights into the phenotypic progression and pathological evolution of ANXA11-associated ALS. Our findings demonstrate that the ANXA11-P36R variant is sufficient to induce a progressive, age-dependent motor neuron disease with inclusion body myopathy in mice. This model recapitulates key features of ALS, including early abnormal protein accumulation, progressive motor neuron loss with neuroinflammation, and late-stage autophagy impairment and mTOR pathway activation (Fig. 9). The extended presymptomatic interval and prolonged survival of this model make it a valuable tool for mechanistic studies and therapeutic interventions in ALS.
Fig. 9
Summary of pathological and phenotypic progression in ANXA11-P36R knock-in ALS mice. The pathological and phenotypic progression in ANXA11-P36R knock-in ALS mice is characterized by early abnormal protein aggregation, motor neuron degeneration, neuroinflammation, and autophagy deficits. At 2 months, abnormal protein aggregates were detected in the central nervous system and muscles, accompanied by spontaneous electromyographic activity. Mutant ANXA11 translocated from the nucleus to the perinuclear region, co-aggregating with p62 and TDP-43. Motor neuron degeneration, including neuronal loss and morphological changes, became pronounced by 9 months, alongside neuroinflammation marked by glial activation and NF-κB translocation. Autophagy deficits, indicated by decreased levels of BECN1 and LC3BII/I, were synchronous with motor decline. Muscle atrophy began to appear from 9-months on and was evident by 12 months, though ANXA11-mutant mice survived beyond 24 months
留言 (0)