Experimental animals
Studies on clinical scores, gait, white matter loss, and in vivo gene expression were all performed on adult female C57BL/6 mice aged 12 − 14 weeks at the beginning of the studies. Time-pregnant CD1 mice and Sprague Dawley rat pups were used to obtain primary astrocyte and primary OPC cultures, respectively. These animals were obtained from Charles River Canada (Charles River, MA, USA). Female mice expressing a neuronal-specific Thy1-eYFP construct on a C57BL/6 background were used to assess axonal injury by the quantification of punctate eYFP fluorescence (The Jackson Laboratory; Stock No: 012708, ME, USA). All studies were performed in accordance with the Canadian Council on Animal Care guidelines and were approved by the Dalhousie University Committee on Laboratory Animals or the Montreal Neurological Institute Animal Care Committee. Mice were housed in the Life Science Research Institute Animal Care Facility on a 12-hr light/dark cycle (7:00 am/7:00 pm); food and water were provided ad libitum. Mice were allowed one week to habituate to the facility prior to experimentation. At the end of all in vivo studies, mice were euthanized with an intraperitoneal (i.p.) injection of 200 µL of sodium pentobarbital (34 mg/mL; Schering Canada, QC, Canada) and intracardially perfused with PBS (10 mL; pH = 7.4).
EAE induction
A peptide fragment corresponding to amino acid sequence 35–55 of mouse myelin oligodendrocyte glycoprotein (MOG35–55; MEVGWYRSPFSRVVHLYRNGK; Gen Script, NJ, USA) was dissolved in phosphate buffered saline (PBS; pH = 7.4) at 3 mg/mL. This solution was then emulsified in complete Freund’s adjuvant (CFA) at a 1:1 ratio by volume. To induce EAE, mice received bilateral 100 µL subcutaneous (s.c.) injections of this final mixture on day post-immunization (DPI) 00. MOG35–55 immunization controls received bilateral s.c. injections of PBS (pH = 7.4) emulsified in CFA at a 1:1 ratio. All mice also received a first 200 µL intraperitoneal (i.p.) injection of pertussis toxin (Sigma-Aldrich, MO, USA) dissolved in water at 1.5 µg/µL on DPI 00 and a second injection on DPI 02.
Clinical scoring
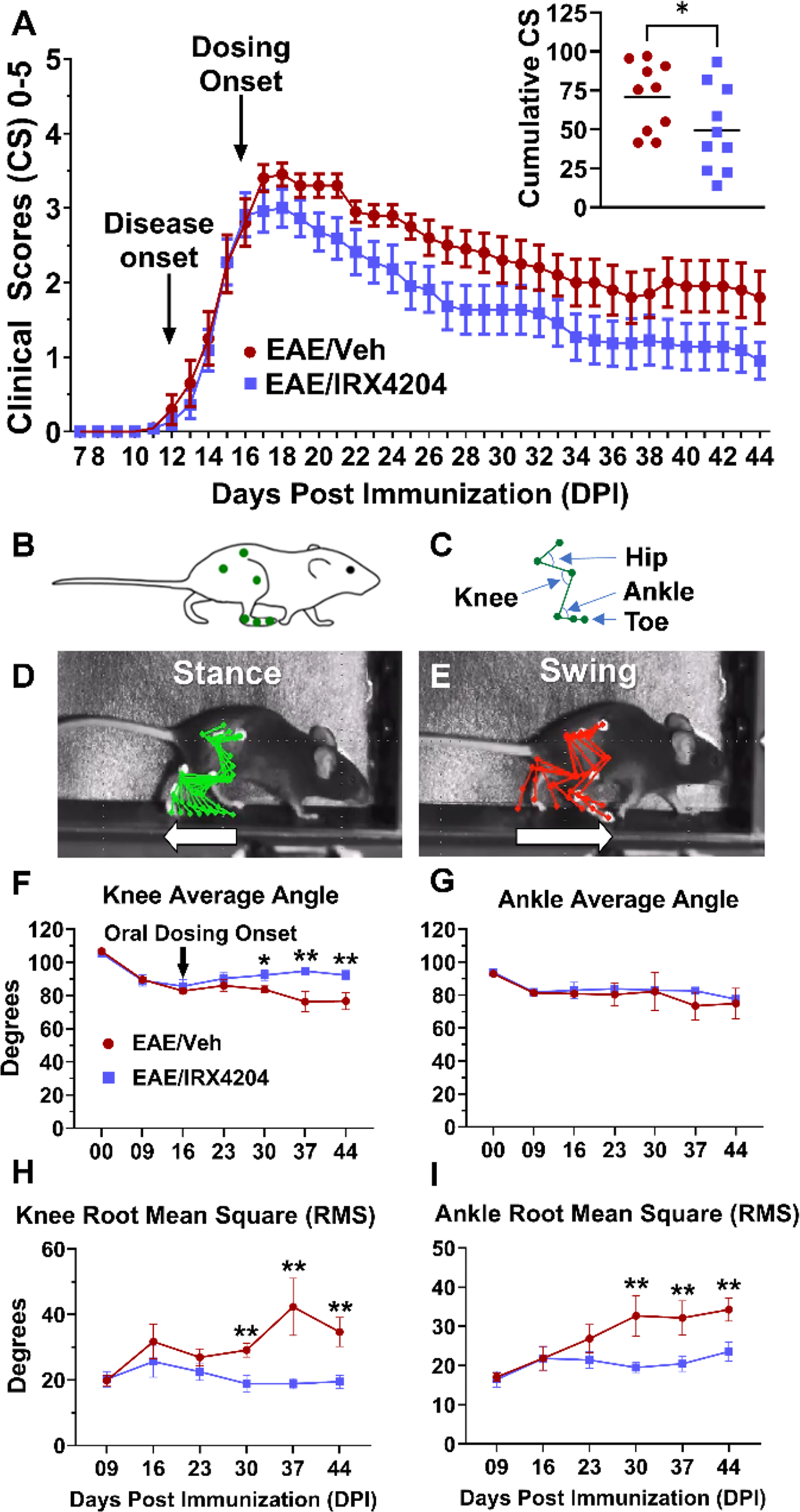
Behavioural deficits associated with increasing disease severity were evaluated using the following ordinal clinical scoring scale: 0, no clinical signs; 0.5, hooked tail; 1, flaccid tail; 1.5, flaccid tail with splay; 2, minor walking deficits, mild ataxia; 2.5, severe walking deficits; 3, dropped pelvis in addition to severe walking deficits, chronic ataxia; 3.5, unilateral hindlimb paralysis; 4, bilateral hindlimb paralysis; 4.5, forelimb paralysis; 5, moribund. Two trained observers began clinical scoring at DPI 07 while blinded to the experimental conditions.
Experimental groups and dosing
For the experimental group designated as EAE/Veh, mice were dosed with 5 mL/kg of vehicle (NEOBEE® 1053; Stepan, Northfield, IL, USA). For the experimental group designated as EAE/IRX4204, mice were dosed with 12 mg/kg IRX4204 (Io Therapeutics Inc., TX, USA). All doses were administered once daily by oral gavage (p.o.). CFA (DPI 16) and EAE (DPI 16) mice from the in vivo gene expression studies were not dosed with vehicle nor with IRX4204 and were euthanized at peak disease (DPI 16) to act as controls for EAE induction.
Kinematic gait analysis
Movements of the right hindlimb were recorded in the sagittal plane as mice walked on a treadmill. A baseline gait recording was taken for each mouse two days prior to the induction of EAE (DPI − 02). Subsequent recordings occurred at weekly timepoints beginning on DPI 09. To ensure that all the mice included were able to walk, kinematic gait analysis was only performed on mice with mild EAE symptoms (clinical score ≤ 2.5). Video recordings were performed using a high-speed camera at a frame rate of 250 frames/s. DeepLabCut software was trained to track the iliac crest, hip, knee, ankle, metatarsophalangeal joint, and toe of mice during each video frame (Fig. 1C and D). This software produced CSV files of X and Y pixel coordinates corresponding to the position of each joint on a frame-by-frame basis. For conversion of pixel distances to centimeter (cm) values, an additional calibration video containing four markers arranged in a 4 cm (height) by 7 cm (width) rectangle was recorded at each timepoint. This calibration recording was taken under the same videographic conditions as those used for all recordings taken at that respective timepoint. One frame from this calibration video was used as input for ImageJ which calculated the conversion coefficient for pixel values to cm values based on the indicated distances between markers. The conversion coefficient and CSV files were used as input into a customized R script. KinemaR used the conversion coefficient to transform the pixilated data into cm values allowing for measurements of joint height and the measurement of hip, knee, and ankle angle in each frame. Phase detection and data normalization were performed by KinemaR as previously described [2, 21, 22, 47]. Briefly, the swing and stance phases (Fig. 1E and F) were detected for each step cycle in a video. Swing and stance phases were each normalized to 100 frames. All of the normalized swing phases and stance phases within a video were then averaged to produce a single representative swing phase and a single representative stance phase of 100 frames each. Together, these comprised the 200-frame representative step cycle for each recording. The representative step cycle was used to calculate average angles for the knee and ankle joints. Joint angles for each frame of the normalized and averaged step cycle were used to calculate the root mean squared (RMS) difference.
Tissue preparation for histology and fluorescence microscopy
EAE/Veh and EAE/IRX4204 mice were euthanized at DPI 44. Mice were then transcardially perfused with PBS followed by 4% paraformaldehyde (PFA; 10 mL; pH = 7.4; Thermo Fisher Scientific, MA, USA) immediately following perfusion with PBS. Spinal cords were dissected from the spinal column by laminectomy and post-fixed in 4% PFA for 24 h. The spinal cords were then transferred to 15% sucrose for 24 h followed by 30% sucrose for 24 h. Next, spinal cord segments L2 − 5 were dissected, embedded in Tissue-Tek® optimal cutting temperature (OCT; Sakura® Finetek, CA, USA) and frozen on dry ice. The embedded samples were then mounted into a Leica CM1950 cryostat set to -18 °C. Serial sagittal sections were cut at a thickness of 30 μm and mounted on Superfrost glass slides (Thermo Fisher Scientific, MA, USA).
Eriochrome cyanine staining
Slides containing sagittal spinal cord sections were dipped into descending concentrations of ethanol at 100%, 90%, and 75% for rehydration. Sections were then rinsed in tap water at room temperature and immersed for 15 min in a 0.16% aqueous solution of Eriochrome Cyanine (EC; Sigma Aldrich, MO, USA) containing 0.4% H2SO4 and 0.4% FeCl3. Sections were then differentiated in 0.5% ammonium hydroxide and counterstained in a 1% solution of Neutral Red (NR; Acros Organics, NJ, USA) for 2 min. Sections were then dipped into a series of increasing concentrations of ethanol at 75%, 90%, and 100% for dehydration prior to being cleared with xylene. Slides were then cover-slipped using Cytoseal 60 (Thermo Fisher Scientific, MA, USA) for imaging using a Zeiss AxioStar Plus (Zeiss, Baden-Württemberg, Germany). Imaging parameters remained the same for all sections. EC stains white matter in blue following differentiation with ammonium hydroxide while NR counterstaining exposes areas where myelin is absent [56]. This dual staining technique was used to quantify white matter loss by measuring the total area of NR staining within white matter regions.
Fluorescence microscopy
Slides containing sagittal spinal cord sections from Thy1-cre/ERT2-eYFP expressing mice were stored at -20 °C in a light-resistant slide box. Sections were brought to room temperature then imaged for endogenous eYFP fluorescence using a Zeiss Axio Imager Z2 with a monochrome camera (Zeiss, Baden-Württemberg, Germany). Bright, punctate labelling in white matter regions of the spinal cord, representative of eYFP aggregation, was used as a marker of axonal transection as described in earlier studies [25]. To discern distinctive areas of bright eYFP labelling in white matter, a threshold was applied to each image. Because the average pixel fluorescence intensity in grey matter was not affected by EAE or treatment groups, threshold values for each image were applied with respect to average grey matter fluorescence. Normalization to grey matter fluorescence provided a final threshold value for each image which accounted for stochastic variation in fluorescence between spinal cord sections. The threshold value was calculated for each image in several steps. First, average pixel fluorescence intensity in healthy appearing white matter was divided by average pixel fluorescence intensity in grey matter in each image. These values were then averaged to obtain a standardized ratio for healthy white matter fluorescence normalized to grey matter fluorescence. Finally, the standardized ratio was multiplied by the average pixel fluorescence intensity of grey matter in each image followed by a factor of 1.5 to obtain a threshold for each image. The percent area of white matter pixels with suprathreshold fluorescence was then calculated over the total white matter area as a measure of percent area axonal transection.
Transmission electron microscopy
EAE/Veh and EAE/IRX4204 mice were euthanized at DPI 30 and intracardially perfused with PBS followed by 2.5% glutaraldehyde (Fisher Scientific, New Jersey, USA) diluted in 0.1 M Sodium Cacodylate Buffer (4 mL; pH = 7.4). Spinal cords were dissected from the spinal column by laminectomy and post-fixed in 2.5% glutaraldehyde for 24 h. Spinal cord segments L3 − 4 were then dissected, rinsed 3 times in 0.1 M sodium cacodylate buffer and fixed in 1% osmium tetroxide for 2 h and, after dehydration, embedded in epon araldite resin. Section 100 nm thick were cut with an ultramicrotome and placed on mesh copper grids, stained with 2% aqueous uranyl acetate, rinsed, and treated with lead citrate, then rinsed again and air dried. Images were captured with a Jeol Jem 1230 transmission electron microscope equipped with a Hamatsu ORCA-HR digital camera at 80 kV and 10 000× magnification. Image analysis for axonal counts and g-ratio calculations was performed using the AxonDeepSeg model model_seg_mouse_axon-myelin_tem [68]. Following automated image analysis, editing of the automated segmentation was performed by an experimenter blinded to experimental conditions to ensure high quality segmentation. Myelinated axons with g-ratios above 0.8 were considered remyelinating axons based on prior characterization in the EAE mouse model [41].
C8-B4 microglial cultures
The C8-B4 microglia cell line (ATCC, VA, USA) was maintained in Dulbecco’s Modified Eagle Medium (DMEM; pH = 7.4; Thermo Fisher Scientific, Cat. # 11995065, MA, USA). For the nitrate release and phagocytosis assays, C8-B4 microglia were seeded in 48-well plates at a density of 150 000 cells/well. To measure gene expression, the cells were seeded in 24-wells plates at a density of 300 000 cells/well. In these assays, the C8-B4 microglial wells were pre-treated with either PBS (pH = 7.4) 0.1, 1, or 10 nM of IRX4204 for 24 h, prior to the addition of lipopolysaccharide (LPS; 100 ng/mL; Sigma-Aldrich, MO, USA) or PBS (pH = 7.4). All assays were performed 24 h following the addition of LPS or PBS (basal conditions).
Microglial nitrate release assay
Cell media from C8-B4 microglial cultures was collected for measurement of nitrate levels using the Greiss reagent in a colorimetric assay, according to manufacturer instructions (Invitrogen™ - G7921, MA, USA). To account for differences in cell abundance between wells, the nitrate levels were normalized to the intensity of crystal violet DNA-staining as measured using the SPECTROstar Nano Absorbance Reader (BMG Labtech, Ortenberg, Germany).
Microglial phagocytosis index
To assess microglial phagocytic activity, uptake of zymosan in these cells was measured in culture. This assay was performed as previously described in Dorighello et al. 2022 [19]. 10 µL of neutral-red stained zymosan (1 × 108 particles/mL) was added to each well and the cells were incubated in 5% (v/v) CO2 for 30 min. Next, the medium was removed, and the cells were fixed with Baker’s solution [4% (w/v) formaldehyde, 2% (w/v) sodium chloride, 1% (w/v) calcium acetate] at 37 °C for 30 min. The macrophages were washed twice with PBS and the neutral-red stain was solubilized with 0.1 mL of acidified alcohol solution [10% (v/v) acetic acid, 40% (v/v) ethanol in distilled water]. After 30 min, the absorbance was monitored at 550 nm using the SPECTROstar Nano Absorbance Reader (BMG Labtech, Ortenberg, Germany). The phagocytosis index was expressed related to crystal violet DNA staining in control wells for normalization to cell abundance.
Astrocyte cultures
Astrocytes were isolated from cerebral cortices of CD1 pups (Charles River Canada, QC, Canada) at postnatal day 0 (P0) mice, as described in Novorolsky 2023 [46]. Following isolation, astrocytes were maintained in astrocyte media [DMEM high glucose (pH = 7.4; Life Technologies, CA, USA) supplemented with 10% FBS (HyClone Laboratories Inc., UT, USA) and 20 µg/mL gentamycin (Thermo Fisher Scientific, MA, USA)] until they reached confluency [day in vitro 7–9 (DIV 7–9)]. These cells were then passed and seeded in 12-well plates at a density of 100 000 cells/well. Plated wells were pre-treated with either PBS (pH = 7.4), 1, 10, or 100 nM of IRX4204 for 24 h prior to the addition of LPS (100 ng/mL; Sigma-Aldrich, MO, USA) or PBS (pH = 7.4). All assays were performed 16 h following the addition of LPS or PBS (basal conditions).
OPC cultures
OPCs were isolated by shake-off from a mixed glial culture derived from the cerebral cortices of P2 rat pups, as previously described [3, 27, 39]. These cells were kept in SATO medium (DMEM supplemented with 5 µg/mL insulin, 50 µg/mL transferrin, 30 nM sodium selenite, 6.3 ng/mL progesterone, 16 µg/mL putrescine, 100 µg/mL penicillin/streptomycin, and 2 mM glutamax) without thyroxine or triiodothyronine at 37 °C in 5% CO2 for 1 day. Finally, OPCs were treated with 1:50,000 DMSO (negative control) or IRX4204 at 0.1, 1, 10 and 100 nM for 1 or 5 days.
OPC Immunocytochemistry
After 2 or 6 DIV cells were fixed for 10 min with 4% PFA in PBS, washed in PBS, and blocked with 2.5% bovine serum albumin and 2.5% horse serum with 0.05% Triton X-100 in PBS for 30 min at room temperature. Cells were incubated overnight at 4 °C with a primary antibody against MBP (1:1000; Aves, #MBP0020) in blocking solution. Upon primary antibody washes, cells were fluorescently labeled with the secondary antibody (1:1000; Alexa Fluor 488, Invitrogen™, MA, USA) diluted in PBS with 2.5% bovine serum albumin and 2.5% horse serum and incubated for 1 h at room temperature. Nuclei were stained with 5 µg/mL Hoechst dye (Sigma-Aldrich, MO, USA) together with the secondary antibody incubation, and three washes with PBS were performed before mounting the coverslips with Dako mounting media. Four randomly chosen areas of each coverslip (4 replicates per condition) were imaged with a 20× objective using a Zeiss Axio Observer.Z1 microscope with an Axiocam 506 camera. Counting MBP positive cells was done manually using the cell counter plugin in ImageJ.
Reverse transcription quantitative polymerase chain reaction (RT-qPCR)
CFA and EAE mice were euthanized at DPI 16 followed by EAE/Veh and EAE/IRX4204 mice at DPI 23 by an overdose injection of pentobarbital. Mice were then intracardially perfused with PBS (10 mL; pH = 7.4). Hydraulic extrusion of the spinal cord was performed using a blunted 18-gauge needle attached to a 10 mL syringe filled with PBS. Spinal cords were then flash-frozen using liquid nitrogen and stored at -80 °C until homogenization in 800 µL pureZOL™ RNA isolation reagent (Bio-Rad Laboratories, Inc., CA, USA) using a bead homogenizer (Benchmark Scientific, NJ, USA). Total RNA was extracted using the Aurum Total RNA Fatty and Fibrous Tissue kit (Bio-Rad Laboratories, Inc., Cat. #732–6870, CA, USA) following the spin protocol as per manufacturer instructions. For C8-B4 microglia and primary astrocyte cultures, total RNA was extracted using the Aurum total RNA minikit (Bio-Rad Laboratories, Inc., Cat. # 732–6820, CA, USA) following the spin protocol as per manufacturer instructions. Concentration and purity of the RNA was estimated spectrophotometrically upon elution using a SPECTROstar Nano spectrophotometer (BMG Labtech, Mandel Scientific Company Inc., ON, Canada). Quality and overall integrity of the isolated total RNA was measured on the Experion™ Automated Electrophoresis System (Bio-Rad Laboratories, Inc., CA, USA) using the Experion RNA StdSens Starter Kit (Bio-Rad Laboratories, Inc., CA, USA). Only samples with RNA Integrity Number (RIN) values of 7.5 or more were considered acceptable and used for further analysis. Generation of complementary DNA (cDNA) was performed according to manufacturer instructions with iScript™ cDNA Synthesis Kit (Bio-Rad Laboratories, Inc., CA, USA) using 850 ng of template from each tissue sample, 600 ng of template from each microglial well, and 180 ng of template from each astrocyte well. cDNA was stored at -20 °C. All qPCR experiments were performed in accordance with the MIQE guidelines on the Bio-Rad CFX96 Real-Time System C1000 Touch Thermal Cycler (Bio-Rad Laboratories, Inc., CA, USA) using the SsoFast EvaGreen Supermix Kit (Bio-Rad Laboratories, Inc., CA, USA) [12]. Each individual gene was optimized for annealing temperature. The RT-qPCR protocol for melt curve analysis used the following conditions: (95 °C for 30 s) + (95 °C x 5 s + 60 °C x 5 s + fluorescence read) x 40 cycles + melt curve analysis. The melting curve program used a 2 s hold time with plate readings for every 0.5 °C increase from 65 °C to 95 °C. ΔΔCq method was used for data analysis using CFX Maestro software (Bio-Rad Laboratories, Inc., CA, USA). HPRT1 and GAPDH were selected as reference genes for the animal and astrocyte studies while GAPDH and β2M were selected as reference genes for the microglial culture studies based on geNorm M values below one for tissue samples and 0.5 for cell cultures. Statistical comparisons were performed using the average value of triplicate technical replicates for all experiments. Primer sequences can be found in supplementary materials.
Statistical analyses
Cumulative clinical scores were calculated for each mouse by adding the daily clinical scores from DPI 16 − 44. For EAE studies, a one-tailed Mann-Whitney U test was used to analyze cumulative clinical scores, percentage of white matter loss, percentage of axonal transection, gene expression in vivo, number of remyelinating axons/µm2, and percentage of remyelinating axons. A two-way repeated measures ANOVA followed by Sidak’s multiple comparisons test was used to analyze gait data at DPI 16, 23, 30, 37, and 44. An ordinary one-way ANOVA followed by Dunnett’s multiple comparisons test was used to analyze the effect of increasing concentrations of IRX4204 on gene expression in vitro, microglial nitrate release, and microglial phagocytosis experiments. A non-parametric Kruskal-Wallis test followed by Dunn’s multiple comparisons test was used to analyze the effect of increasing concentrations of IRX4204 on the proportion of MBP-positive cells to DAPI-positive cells and the effect of increasing concentrations of IRX4204 on the number of DAPI-positive cells per mm2 in OPC cultures. All statistical analyses were performed using GraphPad Prism 8.0.






留言 (0)