
記住我





All animal experiments adhered to guidelines set forth by the Association for Research in Vision and Ophthalmology as well as Animal Research: Reporting of In Vivo Experiments. Animal experiments were approved by the Institutional Animal Care and Use Committees of the University of Pittsburgh and Stanford University. We used both male and female C57BL/6 mice for this research.
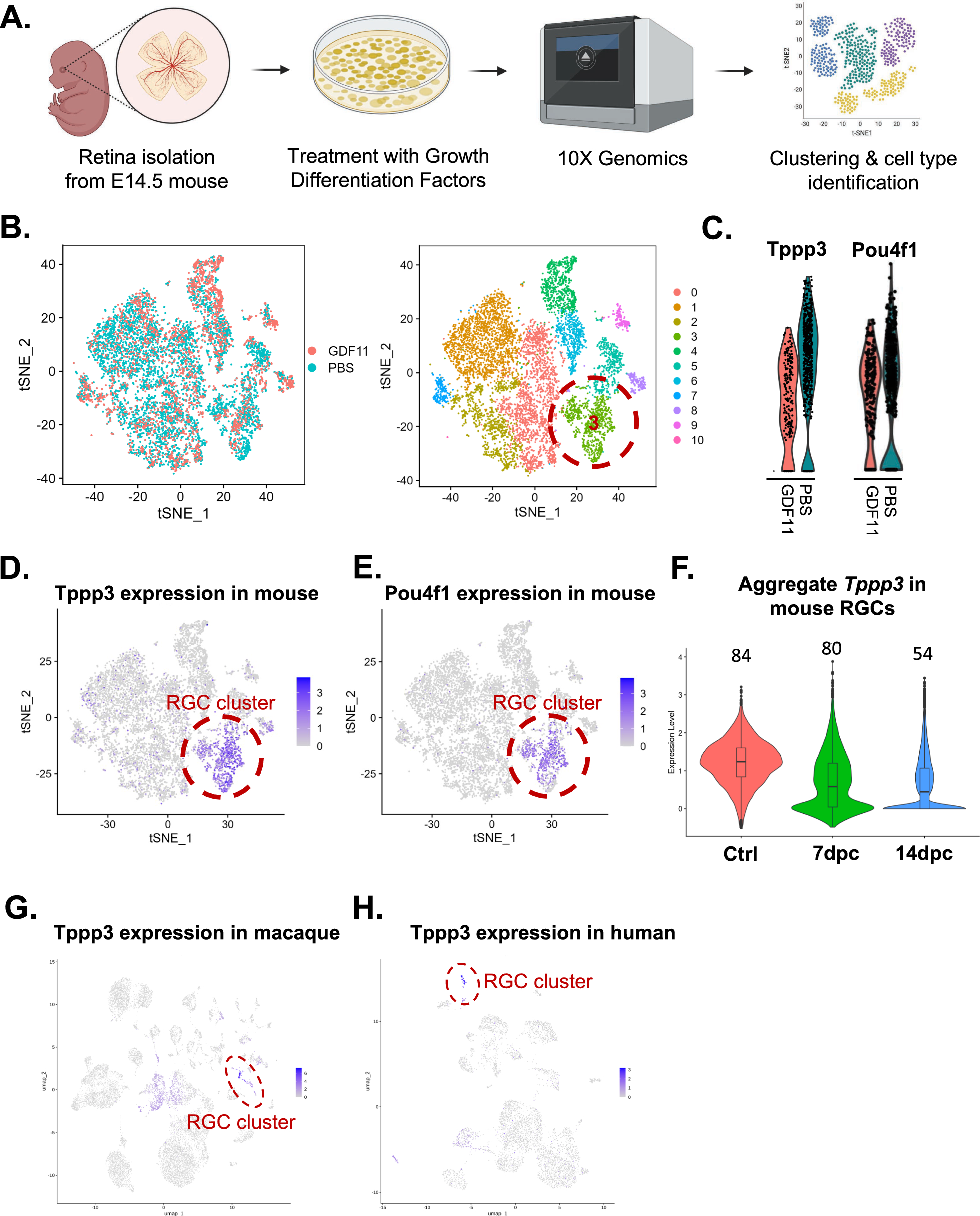
Single-cell RNA sequencingE14.5 mouse retinas were dissociated using papain (#LK003150, Worthington) and re-suspended in retinal progenitor cell (RPC) media. RPC media comprised of glucose (0.6%, Sigma), GS21 (1:100, GlobalStem), Sato supplement (1:100), insulin (5 μg/mL, Sigma), epidermal growth factor (20 ng/mL, Peprotech), fibroblast growth factor (20 ng/mL, Peprotech), penicillin/streptomycin (1%, Thermo Fisher Scientific) suspended in DMEM/F12 medium. The dissociated cells were plated on poly-D-lysine-(PDL)/Laminin-coated dishes. Cells were treated with 50 ng/mL mouse growth differentiation factor 11 (GDF11), or PBS for 5 days. GDF 11 was purchased from R & D systems (#1958-GD-010/CF). Treated cells were then dissociated by Accutase (Innovative Cell Technologies) for 15–20 min on day 6. Cell pellets were filtered through a 40um cell strainer (Falcon, #352,340) and resuspended in PBS with 0.04% BSA. We targeted 10,000 cells for 10X capturing. GEM generation, reverse transcription, cDNA amplification, and library constructions were performed following the manufacturer’s instructions (Chromium Single Cell 3’v1/v2/v3 platform, 10X Genomics, Pleasanton, CA). Samples were sequenced on an Illumina NextSeq 500.
We applied FASTQ with the default parameters, filtering the adaptor sequence and achieving clean data by removing the low-quality reads. Then, we obtained the feature-barcode matrices by aligning reads to the mm10 genome using CellRanger v3.0.0. Seurat analysis was performed in R using Seurat (v3.2.0), ggplot2, and dplyr. We first log normalized the data and identified variable features by scaling gene content by cells. Different treatment datasets (i.e. GDF11, and control) were integrated by identifying ‘anchors’ across datasets; the data was scaled subsequently. Cell clusterings were visualized by tSNE dimensional reduction in Fig. 1. In Fig. 1D, the RGC population was identified by marker gene Pou4f1. Then, the Tppp3 expression level was visualized specifically in the RGC subset (GSE252861).
Fig. 1
Identification of Tppp3 by single-cell RNA sequencing. A Schematic representation of the Experimental design for single-cell RNA sequencing (scRNA-seq) conducted on E14.5 retina samples treated with GDF11 or PBS. B t t-distributed stochastic neighbor embedding (t-SNE) visualization of retinal progenitor cells, with cells color-coded based on their cluster assignments and treatment conditions. Cluster 3 represents the RGC cluster. C GDF11, RGC fate suppressor protein, leads to a reduction in the expression levels of Pou4f1 and Tppp3, specifically within the RGC cluster (cluster 3). This highlights the potential role of Tppp3 in RGC differentiation. D Pou4f1 expression is specifically localized within cluster 3, identified as the RGC-specific cluster. E Tppp3 is also highly expressed within cluster 3. F Violin plot displaying the expression level of Tppp3 from a reanalysis of scRNA data obtained from purified RGCs. The X-axis represents the time points following optic nerve crush, while the numbers above the violin plots indicate the percentage of RGCs expressing Tppp3. 2 weeks after ONC, Tppp3 expression is reduced substantially. Tppp3 is highly expressed within the RGC clusters of G macaque and H humans
In this study, we also re-analyzed previously published scRNA sequencing data sets. scRNA sequencing data were accessed on Gene Expression Omnibus (GEO) under accession numbers GSE199840 (human retina data), GSE161645 (macaque retina data), and GSE137400 (mouse RGC data and ONC). Furthermore, Broad Institute’s Single Cell Portal website was utilized for data visualization of the mouse RGC dataset after ONC- GSE137400.
Adeno-associated virus (AAV) preparationAdeno-associated virus type 2 vectors driving Tppp3 overexpression- CMV > mTppp3(overexpression):P2A:EGFP (AAV2-Tppp3-OE), Tppp3 knockdown CMV > mTppp3(shRNA):P2A:EGFP (AAV2-shTppp3) or cytomegalovirus (CMV) control (AAV2-control and AAV2-shCtrl) were purchased from VectorBuilder. Both the AAV2-control virus and the AAV2-shCtrl virus vector sequences include GFP and are driven by the CMV promoter. AAV2-shCtrl contains a scramble sequence. The plasmids were sent to AAVnerGene for AAV packaging and purification (Rockville, MD, USA). The AAV2 vectors were produced with 293 T cells and purified using CsCl gradient ultracentrifugation. Vector genome concentration was titered by quantitative real-time PCR with iTR primers and digested plasmid as standard (AAV2-Tppp3-OE: 6.92 × 1011 vg/mL, AAV2-control: 8.95 × 1011 vg/mL, AAV2-shTppp3: 7.29 × 1011 vg/mL, AAV2-shCtrl: 5.05 × 1011 vg/mL).
Western blotProtein samples were collected from primary RGCs purified from P2 mouse pups by immunostaining based on the expression of CD90.1 as previously described [4]. The remaining non-RGC cells collected while immunopanning were used as the RGC-depleted population. Samples were collected using Laemmli sample buffer (Sigma-Aldrich, St. Louis, MO, USA). Protein lysates were heated at 100 °C for 10 min, loaded onto precast SDS-PAGE gels (Bio-Rad, Hercules, CA, USA), and run to achieve complete protein separation. A semi-dry blotter from Bio-Rad was used to transfer proteins onto polyvinylidene difluoride membranes. Membranes were blocked for 1 h at room temperature using LI-COR Intercept Blocking Buffer (LI-COR Biosciences, Lincoln, NE, USA).
Membranes were incubated overnight at 4 °C with primary antibodies in LI-COR Intercept Antibody Buffer for immunodetection. Primary antibodies used included those to BRN3A (1:500, MAB1585, Millipore Sigma, Burlington, MA), THY1 (1:500, #5568S, Cell Signaling), RBPMS (1:500, #1832, PhosphoSolutions, Aurora, CO, USA), TPPP3 (1:500, PA5-24,925, Invitrogen, Waltham, MA, USA), and GAPDH (1:500, #2118S, Cell Signaling). Following primary antibody incubation, membranes were incubated with species-specific secondary antibodies linked to near-infrared dyes (#926–68,072, IRDye 680RD donkey anti-mouse; #926–3211, IRDye 800CW goat anti-rabbit; LI-COR Biosciences) at a dilution of 1:10,000 for 4 h at room temperature. Membranes were washed and imaged on a LI-COR Odyssey IR using a linear range detection system.
HeLa cells, obtained from ATCC, were cultured in Eagle's Minimum Essential Medium supplemented with 10% fetal bovine serum, maintained at 37 °C with 5% CO₂. The cells were first transduced with either AAV2-Tppp3-OE or AAV2-control for two days. Following this initial treatment, to test the shRNA transduction efficiency, AAV2-Tppp3-OE-transduced cells were treated with either AAV2-shTppp3 or AAV2-shCtrl, while AAV2-control-transduced cells received AAV2-shCtrl. After treatment, the cells were harvested as previously described and probed for Tppp3 and GAPDH expression.
ImmunostainingEye globes were fixed with 4% paraformaldehyde (PFA) at 4 °C overnight, then incubated in 15% sucrose at 4 °C overnight and 30% sucrose at 4 °C overnight before mounting in Optimal Cutting Temperature mounting medium (Thermo Fisher Scientific, Waltham, MA, USA). 10 μm-thick cryosections were cut from the embedded eye globe. Sections were incubated in blocking buffer containing 5% normal goat serum (NGS) and 0.1% Triton X-100 in PBS for 1 h at room temperature. After three PBS washes, sections were incubated overnight at 4 °C with primary antibodies to mouse Brn3a (1:100, Millipore Sigma, Burlington, MA), mouse anti-β-III-tubulin antibody E7 (1:500, hybridoma from Developmental Studies Hybridoma Bank), and rabbit anti-Tppp3 (1:200, PA5-24,925, Invitrogen).
Following overnight incubation, sections were incubated with secondary antibodies for 4 h at room temperature. We used Alexa Fluor 488 goat anti-rabbit (1:500, #A11034, Life technologies), Alexa Fluor 647 anti-mouse (1:500, A-1235, Life Technologies) and 4′,6-diamidino-2-phenylindole (DAPI) (1:500, #D9542, Sigma-Aldrich). Images were captured on an Olympus Life Science IX83 inverted microscope.
RNAscope in situ hybridization (ISH)In situ detection of Tppp3 mRNA on mouse tissue was performed by a manual method using the RNAscope kit (Advanced Cell Diagnostics) as previously described [11]. Briefly, 12 μm OCT-frozen tissue sections (E12, E14, E18, and P0) were pretreated with hydrogen peroxide, antigen retrieval, and protease application before hybridization with a target probe to mouse Tppp3. Incubation processes were followed by the manufacturer’s instructions. Colorimetric substrate (red) was added to sections and incubated for 10 min at room temperature for observation. Multiple tissues were tested and individual representative sections were shown.
RGC culture and neurite outgrowthPrimary mouse RGCs were purified from postnatal day 2 pups by immunopanning using immobilized antibodies against CD90.1 as previously described [4]. Primary RGCs were plated in a 24-well plate at a density of 2.5 × 103 RGCs/well onto PDL/laminin-coated tissue culture plates. Full Sato medium which included forskolin (5 mm), BDNF (50 ng/mL), and CNTF (10 ng/mL) was used for neurite outgrowth assays. The purity of immunopanned RGCs was validated through immunostaining with Tuj1, Brn3a, and DAPI. As shown in Supplementary Fig. 5, the majority of cells were confirmed to be RGCs. For viral transduction, AAV2-Tppp3-OE or AAV2 control was added to the culture medium at a multiplicity of infection of ∼105 vg/cell 24 h. Similarly, AAV2-shTppp3 or AAV2-shCtrl were added to the culture medium. The virus was removed after 24 h and RGCs were incubated for 2 days in fresh medium.
For immunostaining, RGCs were fixed in 4% PFA directly added to the culture medium (2% final PFA dilution) for 20 min. Neurites were blocked in 5% NGS and 0.1% Triton X-100 in PBS for 1 h at room temperature. Rabbit Tuj-1 (1:500; #5568S, Cell Signaling) was used to stain neurites overnight and was visualized with an Alexa Fluor 555-conjugated Goat Anti-rabbit IgG antibody (1:500, # A-21428, Life Technologies) for 4 h at room temperature. Neurite outgrowth was measured using an Olympus Life Science IX83 Inverted Microscope. Each image was taken at the same intensity and 10X magnification. For each treatment, ~ 30 cells were averaged per condition for each experiment. The total length of neurites per cell was measured using ImageJ Simple Neurite Tracer. All imaging and quantification were conducted in a blinded manner to eliminate bias.
Optic nerve crush (ONC)Adeno-associated virus type 2 vectors driving Tppp3 overexpression (AAV2-Tppp3-OE) or cytomegalovirus control (AAV2-control) were intravitreally injected into the left eye two weeks before ONC. ONC was performed as previously described [9]. Ketamine/xylazine anesthesia was administered to 8–10-week-old mice. The left eye was subjected to ONC—the outer canthus was exposed, and the optic nerve was pinched for 7 s using Dumont #5 self-closing forceps (Fine Science Tools, Foster City, CA, USA), ~ 1.5 mm behind the globe. The right eye was left uninjured to serve as a control. At 12 days after ONC, 2 µL of cholera toxin subunit B (CTB)-conjugated Alexa Fluor 555 (CTB-555) (2 μg/μL, #C22843, Invitrogen) were intravitreally injected as an anterograde tracer. Animals were euthanized 14 days after ONC and perfused with 4% PFA before collecting optic nerves and retinas.
Quantitative real-time PCR (qPCR)We isolated total RNA from retinal tissues collected from E12-P0, two days after ONC and two weeks using a Qiagen RNeasy Mini Kit per the manufacturer's protocol (Qiagen, Hilden, Germany). Reverse transcription of RNA (500 ng) was performed using the Bio-Rad iScript cDNA Synthesis Kit. qPCR was conducted using either Taqman master mix or Bio-Rad iTaq Universal SYBRGreen Supermix per the manufacturer's instructions. SYBR green primers for Bmp4 and Gapdh were purchased from Integrated DNA Technologies (Coralville, IA, USA). All experiments were performed in triplicate to ensure accuracy and reproducibility.
RGC survival analysisRGC survival was evaluated according to an established protocol [10]. First, retinas were dissected and fixed in 4% PFA for 1 h. To ensure complete permeabilization, retinas were treated with a solution containing 3% Triton X-100 (Sigma-Aldrich) and 1.5% Tween 20 (Sigma-Aldrich) for 1 h. Blocking was performed using 10% normal goat serum in PBS for 1 h. Retinas were incubated overnight at 4 °C with a rabbit polyclonal anti-RBPMS primary antibody (1:500, #1830, PhosphoSolutions). Following three washes with PBS (10 min each), retinal flatmount samples were incubated overnight with Alexa Fluor 647-goat anti-rabbit secondary antibody (1:500, #A21244, Life Technologies). After two additional washes (10 min each), samples were stained with DAPI (1:5000 in PBS) for 15 min. For preservation, samples were sealed under 1.5-mm coverslips using an anti-fade mounting medium (ProLong Gold, Life Technologies). Samples were imaged on a Zeiss fluorescence microscope (Oberkochen, Germany). Each retina was divided into four quadrants, and one random digital micrograph was captured from each peripheral area located 3 mm from the optic nerve head. RBPMS-positive (RBPMS +) cells were manually counted in a masked manner. Results are presented as cells per square millimeter.
Regenerative axon countingFor axon counting, optic nerves were collected two weeks after ONC, fixed in PFA for 1 h at room temperature, and washed in PBS. Optic nerves were immersed in 15% sucrose at 4 °C overnight, followed by 30% sucrose at 4 °C overnight before mounting in Optimal Cutting Temperature mounting medium (Thermo Fisher). Cryosections 10-μm thick were prepared for both the optic nerve and retina. Optic nerve sections were imaged and analyzed as described previously [9]. The number of CTB-positive axons (CTB +) within every 200 μm, 600 μm, 1000 μm and 1600 μm from the injury site were manually counted until the end of the longest regenerating axons. The total number of CTB + axons per optic nerve was calculated using an established formula [23]. All imaging and quantification were conducted in a masked manner.
RNA sequencingRNA sequencing was performed to investigate gene expression profiles in the injured retina two days after ONC. Mouse retinas treated with either AAV2-Tppp3-OE (n = 4) or AAV2-CMV (n = 3) were dissected, and total RNA was extracted from the injured retinas. Total RNA was extracted from frozen retina samples using a Qiagen RNeasy Mini Kit per the manufacturer’s instructions. Sample preparation was conducted in triplicate to ensure robustness and reproducibility.
Azenta Life Sciences (South Plainfield, NJ, USA) conducted sample, library preparation, and quality control analyses. Briefly, RNA samples were quantified using a Qubit 2.0 fluorometer (Life Technologies), and RNA integrity was checked using a TapeStation 4200 (Agilent Technologies, Palo Alto, CA, USA). ERCC RNA Spike-In Mix Kit (#4,456,740, Thermo Fisher Scientific) was added to normalized total RNA before library preparation following the manufacturer’s protocol. RNA sequencing libraries were prepared using the NEBNext Ultra II RNA Library Prep Kit for Illumina per the manufacturer’s instructions (New England Biolabs, Ipswich, MA, USA). Sequencing libraries were validated on an Agilent TapeStation and quantified using a Qubit 2.0 fluorometer (Thermo Fisher Scientific) as well as by quantitative PCR (KAPA Biosystems, Wilmington, MA, USA). Sequencing libraries were clustered on one flow cell lane. After clustering, the flow cell was loaded on an Illumina 4000 or equivalent instrument according to the manufacturer’s instructions (Illumina, San Diego, CA, USA). Samples were sequenced using a 2 × 150 bp paired-end configuration. Raw sequence data (.bcl files) were converted into FASTQ files and de-multiplexed using Illumina bcl2fastq 2.17 software. One mismatch was allowed for index sequence identification.
The resulting FASTQ files were analyzed using CLC Genomics Workbench v22 from Qiagen Digital Insights. Raw sequencing reads were imported into the software for analysis, including identifying differentially expressed genes between AAV2-Tppp3-OE and AAV2-CMV groups two days after ONC. The imported reads underwent quality checks, adaptor sequence trimming, and alignment to the GRCm39/mm39 version of the mouse reference genome using default settings. Quality checks were performed on mapped reads as well.
To determine the differential expression of genes between groups, a significance threshold of p ≤ 0.05 and fold-change > 2 were used. Adjusted p-values were not used to compute differentially expressed genes in this analysis. Volcano plots were generated using CLC Genomics Workbench v22 to visualize results. Pathway enrichment analyses were conducted using the NIH Database for Annotation, Visualization, and Integrated Discovery 2021 with the Kyoto Encyclopedia of Genes and Genomes database. RNA sequencing data generated from this analysis have been deposited in Gene Expression Omnibus (GEO) under accession number GSE24244756.
Statistical analysisStatistical analysis was performed by calculating the mean ± standard error of the mean (SEM) of at least three independent experiments. The number of mice used in each experiment is indicated in the figure legends. One-way ANOVA followed by post hoc t-tests with Tukey’s correction and/or unpaired t-tests was used for data analysis, considering p < 0.05 as significant. Graphs were created using Prism 9 software (GraphPad, La Jolla, CA, USA).
留言 (0)