Chemicals
Unless otherwise stated, all chemicals and reagents used were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Oocyte recovery and in vitro maturation
Porcine ovaries were obtained from a local slaughterhouse, placed in 0.9% physiological saline at 38.5 °C, and transported to the laboratory within 2 h. Using a syringe, COCs were collected from the surface of the ovarian follicles (3–7 mm in diameter), and those containing three or more intact cumulus layers were selected. The COCs were rinsed three times with an in vitro maturation (IVM) medium composed of tissue culture medium 199, 10 ng/mL epidermal growth factor, 25 µM β-mercaptoethanol, 0.57 mM cysteine, 10% porcine follicular fluid, 10 IU/mL pregnant mare serum gonadotropin (PMSG; ProSpec, Rehovot, Israel), and 10 IU/mL human chorionic gonadotropin (hCG; ProSpec). Approximately 50 COCs were incubated in IVM medium with paraffin oil (Junsei, Tokyo, Japan) for 22 h at 38.5 °C under 5% CO2. Thereafter, the COCs were transferred to PMSG- and hCG-free IVM medium and incubated for another 22 h at 38.5 °C under 5% CO2.
Chemical treatment
A stock solution of 20 mM RO (Selleckchem, Houston, TX, USA) was prepared with dimethylsulfoxide and diluted in IVM medium to final concentrations of 0, 5, 10, and 20 µM.
Assessment of the cumulus expansion and nuclear maturation of oocytes
A total of 1000 COCs was used in five independent replicates. After 44 h of IVM, cumulus expansion was evaluated on the basis of the morphology of the COCs and classified into four levels: degree 1 (cumulus cells that are spherical and compacted with minimal observable expansion), degree 2 (cumulus cells that are partially expanded with an outer layer), degree 3 (cumulus cells that are mostly expanded except for the corona radiata), and degree 4 (cumulus cells that are fully expanded). To determine the nuclear maturation of the oocytes after 44 h of IVM, the COCs were placed in a denuding medium containing Dulbecco’s phosphate-buffered saline (DPBS; Gibco, Grand Island, NY, USA), 100 μg/mL penicillin G, 75 μg/mL streptomycin sulfate, and 0.1% hyaluronidase. After gently pipetting the COCs, the denuded oocytes were observed under a microscope (Nikon Corp., Tokyo, Japan) and classified as immature (without polar body extrusion), metaphase II (MII; with polar body extrusion), or degenerate.
Parthenogenetic activation and in vitro culture
A total of 631 MII oocytes was used in five independent replicates. For parthenogenetic activation, MII oocytes were treated with 15 μM of ionomycin in DPBS (Gibco) supplemented with 60 μg/mL gentamicin sulfate, 75 μg/mL streptomycin sulfate, and 4 mg/mL bovine serum albumin (BSA) for 5 min in the dark. After activation, the oocytes were incubated in in vitro culture medium (PZM-3 medium containing 4 mg/mL BSA) supplemented with 5 µg/mL cytochalasin B and 2 mM 6-dimethylaminopurine for 4 h at 38.5 °C under 5% CO2. Subsequently, the oocytes were transferred to fresh in vitro culture medium and cultured for 6 days at 38.5 °C under 5% CO2.
Immunocytochemistry
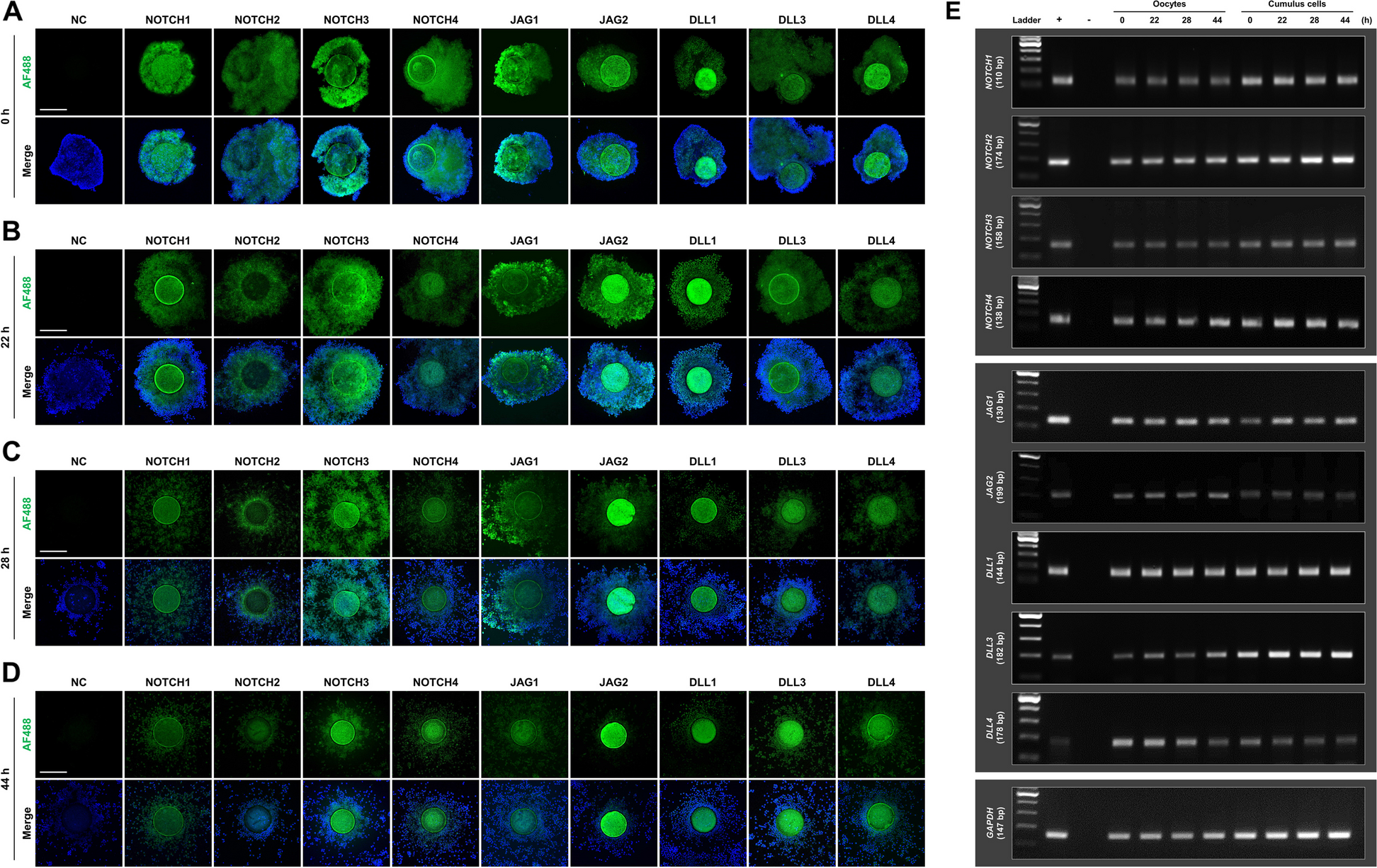
For NOTCH1 staining, in total, 49, 50, 48, and 48 COCs were sampled at different time points (0, 22, 28, and 44 h of IVM, respectively) and used in four independent replicates. For NOTCH2 staining, in total, 43, 48, 46, and 47 COCs were sampled at different time points (0, 22, 28, and 44 h of IVM, respectively) and used in four independent replicates. For NOTCH3 staining, in total, 35, 36, 37, and 35 COCs were sampled at different time points (0, 22, 28, and 44 h of IVM, respectively) and used in four independent replicates. For NOTCH4 staining, in total, 45, 43, 45, and 43 COCs were sampled at different time points (0, 22, 28, and 44 h of IVM, respectively) and used in four independent replicates. For JAG1 staining, in total, 43, 43, 40, and 44 COCs were sampled at different time points (0, 22, 28, and 44 h of IVM, respectively) and used in four independent replicates. For JAG2 staining, in total, 49, 46, 47, and 48 COCs were sampled at different time points (0, 22, 28, and 44 h of IVM, respectively) and used in four independent replicates. For DLL1 staining, in total, 39, 38, 38, and 38 COCs were sampled at different time points (0, 22, 28, and 44 h of IVM, respectively) and used in three independent replicates. For DLL3 staining, in total, 42, 41, 38, and 38 COCs were sampled at different time points (0, 22, 28, and 44 h of IVM, respectively) and used in three independent replicates. For DLL4 staining, in total, 42, 41, 42, and 39 COCs were sampled at different time points (0, 22, 28, and 44 h of IVM, respectively) and used in three independent replicates. For GDF9 staining, a total of 40 MII oocytes was used in three independent replicates. For BMP15 staining, a total of 40 MII oocytes was used in three independent replicates. For CDX2 staining, a total of 66 blastocysts was used in three independent replicates. For α-Tubulin staining to evaluate the proportions of Pro-MI and MI stage oocytes, a total of 348 oocytes was used in five independent replicates. For α-Tubulin staining to evaluate the chromosome and spindle morphology, a total of 144 oocytes was used in four independent replicates. For BUBR1 staining, a total of 96 oocytes was used in three independent replicates. For Ac-Tubulin staining, a total of 70 oocytes was used in three independent replicates. For HES1 staining, a total of 60 COCs was used in three independent replicates. For HES2 staining, a total of 42 COCs was used in three independent replicates. For HEY1 staining, a total of 54 COCs was used in three independent replicates. For HEY2 staining, a total of 60 COCs was used in three independent replicates. COCs, oocytes, and blastocysts were washed three times in DPBS (Welgene, Daegu, Republic of Korea) supplemented with 0.1% polyvinyl alcohol (PBS-PVA) and then fixed overnight in 4% paraformaldehyde at 4 °C. After washing three times with PBS-PVA, the respective cells were incubated in PBS supplemented with 1% Triton X-100 for 1 h at ambient temperature. Then, the permeabilized samples were washed three times with PBS-PVA. Subsequently, the cell samples were placed in PBS-PVA containing 1 mg/mL BSA (PBS-PVA-BSA) at ambient temperature for 1 h to block nonspecific binding. To stain for caudal type homeobox 2 (CDX2), blastocysts were further incubated in PBS containing 10% normal goat serum for 1 h. The cell samples were incubated with primary antibodies overnight at 4 °C. The specific antibodies used are listed in Supplementary Table 1. After washing three times with PBS-PVA-BSA, the samples were treated with Alexa Fluor 488-labeled goat anti-rabbit, goat anti-mouse, or donkey anti-goat IgG secondary antibodies (Invitrogen, Waltham, MA, USA) for 1 h at ambient temperature. Following this, the samples were washed three times with PBS-PVA-BSA and mounted on glass slides with mounting solution containing 1.5 µg/mL 4′,6-diamidino-2-phenylindole (DAPI; Vector Laboratories, Newark, CA, USA). Cell fluorescence was observed using a laser scanning confocal fluorescence microscope (LSM700; Carl Zeiss, Oberkochen, Germany) or a fluorescence microscope (DMi8; Leica Microsystems, Wetzlar, Germany), and the fluorescence intensity was analyzed using ImageJ software after normalization through subtraction of the background intensity to that of control.
Terminal deoxynucleotidyl transferase-mediated dUTP-digoxygenin nick end-labeling assay
A total of 80 blastocysts was used in five independent replicates. The In Situ Cell Death Detection Kit (Roche, Basel, Switzerland) was used for the terminal deoxynucleotidyl transferase-mediated dUTP-digoxygenin nick end-labeling (TUNEL) assay. Blastocysts were fixed overnight in 4% paraformaldehyde at 4 °C, then washed three times with PBS-PVA, and subsequently permeabilized in DPBS with 1% (v/v) Triton X-100 for 1 h at ambient temperature. Thereafter, the blastocysts were washed three times with PBS-PVA and incubated with fluorescein-conjugated dUTP and terminal deoxynucleotidyl transferase for 1 h at 38.5 °C. After the incubation, the blastocysts were washed three times with PBS-PVA and then mounted on slides with mounting solution containing 1.5 µg/mL DAPI (Vector Laboratories). Cell fluorescence was observed using a fluorescence microscope (DMi8; Leica Microsystems).
Reverse transcription and quantitative polymerase chain reactions
Poly(A) mRNAs were isolated from cumulus cells, oocytes, and blastocysts using the Dynabeads mRNA Direct Kit (Invitrogen) following the manufacturer’s protocol and then reverse transcribed using the PrimeScript™ RT Reagent Kit with gDNA Eraser (Takara Bio Inc., Shiga, Japan) according to the manufacturer’s protocol. A total of 50 COCs or 10 blastocysts were sampled in each batch for Poly(A) mRNA extraction and used in three independent replicates. For the reverse transcription polymerase chain reactions (RT-PCR), a SureCycler 8800 system (Agilent Technologies, Santa Clara, CA, USA) was used: 3 µl cDNA, 1 µl (5 pM) forward primer, 1 µl (5 pM) reverse primer, 10 µl ExPrime Premix (GeNet Bio, Daejeon, Republic of Korea), and 5 µl of Nuclease-free water (Invitrogen). The PCR products were electrophoresed for 30 min at 110 V using a JY600 Basic Power Supply (JUNYI, Beijing, China). The results were visualized and photographed using a gel imaging system (Fusion SOLO S; Vilber, Marne La Vallée, France). SYBR Premix Ex Taq (TaKaRa Bio) and the AriaMx Real-time PCR system (Agilent) were used for the quantitative PCR: 3 µl cDNA, 1 µl (5 pM) forward primer, 1 µl (5 pM) reverse primer, and 5 µl SYBR Premix Ex Taq (TaKaRa Bio). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as the internal standard for comparative analyses. The specific primers used for the PCRs are listed in Supplementary Table 2.
Chromosome spreading
A total of 226 oocytes was used in six independent replicates. To spread the chromosomes, the oocytes were treated with acidic Tyrode’s solution to remove the zona pellucida. After a brief recovery in fresh medium, the oocytes were fixed and spread on glass slides using 1% paraformaldehyde in distilled water (pH 9.2) containing 0.15% Triton X-100 and 3 mM dithiothreitol. The slide samples were dried slowly for several hours at ambient temperature in a humid chamber, following which they were covered with a blocking solution for 1 h at ambient temperature. Then, the cells were incubated first with BUBR1 antibody (Abcam, Cambridge, UK) overnight at 4 °C and then with Alexa Fluor 488-labeled goat anti-rabbit secondary antibody for 1 h at ambient temperature. Finally, the DNA was stained by dropping 10 μL of mounting solution containing 1.5 µg/mL DAPI (Vector Laboratories) onto the cells. Cell fluorescence was examined using a laser scanning confocal fluorescence microscope (LSM700; Carl Zeiss).
Analysis of actin filaments
A total of 279 MII oocytes was used in three independent replicates. MII oocytes were fixed overnight with 4% paraformaldehyde at 4 °C, then incubated in DPBS containing 1% Triton X-100 for 1 h at ambient temperature, and subsequently blocked with 2 mg/mL BSA in PBS-PVA for 1 h at ambient temperature. Then, the oocytes were treated with 10 μg/mL phalloidin-tetramethylrhodamine B isothiocyanate for 2 h at ambient temperature. After washing in PBS-PVA, the oocytes were mounted onto glass slides with a mounting solution containing 1.5 µg/mL DAPI and observed using a laser scanning confocal fluorescence microscope (LSM700; Zeiss).
Analysis of mitochondria and endoplasmic reticula
For MitoTracker staining, a total of 156 MII oocytes was used in five independent replicates. For JC-1 staining, a total of 96 MII oocytes was used in three independent replicates. For ER-Tracker staining, a total of 236 MII oocytes was used in nine independent replicates. To assess the mitochondrial and endoplasmic reticulum (ER) activities, MII oocytes were incubated in IVM medium containing 200 nM MitoTracker Red CMXRos (Invitrogen), JC-1 (1:100) (Cayman Chemical, Ann Arbor, MI, USA), or 1 µM ER-Tracker (Invitrogen) for 1 h. Subsequently, the oocytes were washed three times with PBS-PVA (10 min each time) and then fixed in 4% paraformaldehyde for 2 h at 38.5 °C. Cell fluorescence was observed immediately using a fluorescence microscope (DMi8; Leica Microsystems). To determine mitochondrial and ER distribution, stained and fixed oocytes were washed three times in PBS-PVA and then mounted onto glass slides using mounting solution containing 1.5 µg/mL DAPI (Vector Laboratories). Cell fluorescence was observed immediately under a fluorescence microscope (DMi8; Leica Microsystems), and the fluorescence intensity was analyzed using ImageJ software after normalization through subtraction of the background intensity to that of control.
Analysis of ATP and cytoplasmic and mitochondrial calcium concentrations
For ATP staining, a total of 80 MII oocytes was used in three independent replicates. For Fluo-3 staining, a total of 100 MII oocytes was used in four independent replicates. For Rhod-2 staining, a total of 160 MII oocytes was used in five independent replicates. MII oocytes were washed three times with PBS-PVA and then fixed overnight in 4% paraformaldehyde at 4 °C. To stain ATP, fixed oocytes were washed three times with PBS-PVA and then incubated in PBS-PVA containing 500 nM BODIPY FL ATP (Molecular Probes, Eugene, OR, USA) for 1 h in the dark at ambient temperature. Subsequently, the oocytes were washed three times with PBS-PVA and then mounted onto glass slides with a mounting solution containing 1.5 µg/mL DAPI. To stain cytoplasmic and mitochondrial calcium, the fixed oocytes were washed three times with PBS-PVA and then incubated in PBS-PVA containing 10 μM Fluo-3 (Invitrogen) and 5 μM Rhod-2 (Invitrogen) for 1 h in the dark at ambient temperature. Sample fluorescence was observed using a fluorescence microscope (DMi8; Leica Microsystems), and the fluorescence intensity was analyzed using ImageJ software after normalization through subtraction of the background intensity to that of control.
Statistical analysis
All statistical analyses were performed using SigmaStat software (SPSS Inc., Chicago, IL, USA). Each experiment was conducted at least three times. One-way analysis of variance was used for comparisons among three or more groups, followed by Tukey’s post-hoc test. Student’s t-test was used to compare the scores between two groups. The results are presented as the mean ± standard error of the mean. Differences were considered statistically significant at a P-value of less than 0.05.





留言 (0)