Sample preparationHuman specimens
A total of 20 kidney tissue samples were collected from participants diagnosed with renal tumors at the First Affiliated Hospital of China Medical University. Exclusion criteria included the absence of prior radiotherapy or chemotherapy before nephrectomy, as well as the presence of systemic dysfunctions [49]. Informed consent was obtained from all participants prior to the study commenced. Given the increased risk of end-stage renal disease and drug-induced nephrotoxicity in individuals over 65 years old [50], participants were divided into two groups based on age (young: < 65 years old; aged: ≥ 65 years old). The clinical characteristics of the participants are presented in Additional file 2: Table S1.
Animal experimental design
All animal experiments were conducted in the SPF (Specific Pathogen-Free) animal laboratory of China Medical University following Guidelines outlined in the NIH guide for the Care and Use of Laboratory Animals. C57BL/6 J male mice were sourced from Charles River Laboratories and housed in a controlled environment with a standard light/dark cycle, along with unrestricted access to water and a chow diet. Tissue samples from the natural aging model were collected at 6 months (n = 8) and 24 months (n = 8), referred to as the 6-month-old and 24-month-old groups, respectively.
The accelerated aging model was developed by administrating D-gal into C57BL/6 mice, thereby simulating the natural aging process in rodents. D-gal activates senescence-related signal pathways by reacting with free amines of amino acids, forming advanced glycation end products and resulting in the formation of advanced glycation end products and the subsequent induction of oxidative stress and inflammation. The experimental strategy was illustrated in Fig. 2A. Briefly, mice received subcutaneous injections of D-gal at 500 mg kg − 1 day − 1 from 8 to 16 weeks. At 9 weeks, adeno-associated viruses (Syngentech, Beijing, China) containing GLIS1 expression vector (AAV2/9-GLIS1) or METTL3 expression vector (AAV2/9-METTL3) were injected into the renal pelvis of D-gal-treated mice. A total volume of 100 μL pAAV2/9-GLIS1-FLAG (NM_147221.2) or pAAV2/9-METTL3-FLAG and an empty vector (1 × 1012 virus genome/ml) were injected into the renal pelvis of mice. Three groups were established as follows: (1) control group (n = 8); (2) D-gal treated group with AAV-GLIS1/AAV-METTL3 (n = 8); (3) D-gal treated group with AAV-vector (n = 8).
Cell culture and treatment
The human kidney TEC line (HK-2) was obtained from the American Type Culture Collection and cultured in DMEM/F12 medium supplemented with 10% fetal bovine serum (FBS) and 1% antibiotic–antimycotic at 37 °C. The culture medium was replaced every three days until the cells reached 90% confluence. To simulate kidney aging, HK-2 cells were treated with D-gal (100 mM) for 72 h, creating the D-gal-treated HK-2 group. Alternatively, HK-2 cells were treated with 25 μM MK-886 or 40 μM etomoxir for 24 h to investigate the roles of peroxisome proliferator-activated receptor alpha (PPARα) and carnitine palmitoyltransferase 1A (CPT1A).
The human over-expressed GLIS1 gene was amplified and subcloned into the eukaryotic expression vector pcDNA3.1. Specific target sequences for GLIS1 siRNA, siFTO, siMETTL3, siYTHDF1, and siACOX1 (Syngentech, China) were transfected into target cells using Lipofectamine 3000 (Invitrogen) according to the manufacturer’s instructions. A scrambled sequence served as the control. The siRNA sequences are listed in Additional file 2: Table S2.
Histological and immunohistochemical (IHC) staining
Human and mouse kidney tissues were cut into 3 μm-thick sections. These samples were stained with Masson and periodic acid-Schiff (PAS) staining and observed under a microscope. For semi-quantitative analysis, the glomerulosclerosis index (GSI) was recorded on a scale from 0 to 4 (0: no lesion; 1: mild sclerosis, 0–25% of the glomerulus affected; 2: moderate sclerosis, 25–50% of the glomerulus affected; 3: severe sclerosis, 50–75% of the glomerulus affected) [1, 2]. Masson staining was used to assess interstitial fibrosis in kidney tissues, with morphometric analysis with scores as follows: 0: < 5% affected interstitial area; 1: 5–25% affected interstitial area; 2: 25–50% affected interstitial area; 3: > 50% affected interstitial area.
IHC was performed according to the manufacturer’s instructions. Specific primary antibodies targeting GLIS1 (Proteintech 23,138–1-AP), P16INK4A (Thermo Fisher Scientific Cat# MA5-17,142), γ-H2AX (Proteintech 10,856–1-AP), FN (Abcam ab2413), α-SMA (Abcam ab5694), PPARα (Novus NB300-537), ACOX1 (Novus NBP1-80950), CPT1A (Abcam ab234111), HK2 (Sigma SAB2108077), PDK1 (Sigma SAB4502160), METTL3 (Abcam ab195352), and YTHDF1 (Abcam ab252346) were incubated overnight at 4 °C. The sections were then incubated with secondary antibodies for 1 h at room temperature. Afterward, the samples were stained with diaminobenzidine (DAB) and counterstained with hematoxylin, followed by visualization using a microscope.
Transmission electron microscope (TEM) observation
Serial Sects. (80 nm) of the samples were prepared using an ultramicrotome and stained with uranyl acetate and lead citrate. After rinsing and vacuum-drying at room temperature, the ultrastructure of the samples, particularly foot process effacement and GBM, were observed using TEM. For semi-quantitative analysis, ImageJ software was employed to measure the thickness of GBM.
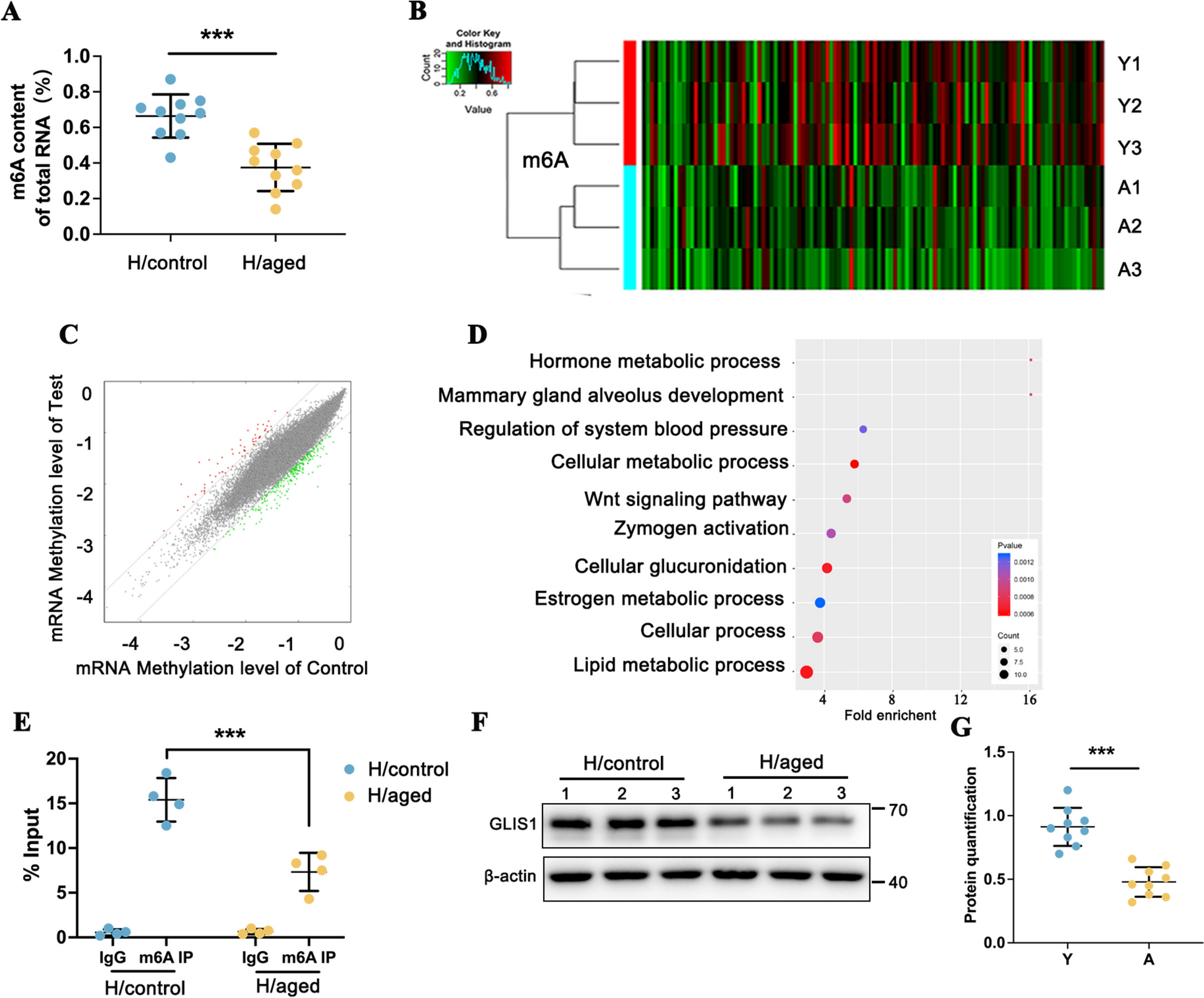
Quantification of m6A modifications
m6A modification was quantified using the EpiQuik m6A RNA Methylation Quantification Kit (Colorimetric). Total RNA was isolated from human kidney tissues using TRIzol solution, and the concentration was determined with a spectrophotometer. A total of 200 ng of RNA was coated on the assay well at 37 °C for 90 min, followed by the addition of capture antibody solution and detection antibody solution. m6A methylation levels were quantified at a wavelength of 450 nm.
Western blot
Protein lysates from kidney tissues and cultured cells were prepared, electrophoresed, and transferred to PVDF membranes. The membranes were blocked with 5% non-fat milk for 1 h at room temperature, followed by overnight incubation with primary antibodies: GLIS1 (Proteintech 23,138–1-AP), P16INK4A (Thermo Fisher Scientific MA5-17,142), γ-H2AX (Proteintech 10,856–1-AP), FN (Abcam ab2413), α-SMA (Abcam ab5694), collagen type I (COL I) (Abcam ab260043), PPARα (Novus NB300-537), ACOX1 (Abcam ab184032), CPT1A (Abcam ab234111), HK2 (Abcam ab209847), PDK1 (Abcam ab110025), METTL3 (Abcam ab195352), METTL14 (Sigma HPA038002), WTAP (Proteintech 10,200–1-AP), FTO (Proteintech 27,226–1-AP), ALKBH5 (Proteintech 16,837–1-AP), YTHDF3 (Proteintech 25,537–1-AP), and YTHDF1 (Abcam ab252346) overnight at 4 °C. Afterward, a secondary antibody was applied for 1 h at room temperature, and the blots were visualized using enhanced chemiluminescence (ECL).
m6A-mRNA epitranscriptomic microarray analysis
Human m6A epitranscriptomic microarray and mRNA microarray analyses were performed based on Arraystar’s standard protocols. Total RNA was immunoprecipitated with an anti-m6A antibody. The immunoprecipitated (IP) and supernatant (Sup) RNA were labeled with Cy5 and Cy3, respectively. The RNAs were then hybridized to the Arraystar Human mRNA Epitranscriptomic Microarray (8 × 60 K, Arraystar). Raw intensities of IP and Sup were normalized using the average of log2-scaled Spike-in RNA intensities. The raw data have been uploaded to the Gene Expression Omnibus database under the accession number GSE232249.
RT-qPCR
The extracted RNAs from kidney tissues or cultured cells were reversely transcribed, and RT-qPCR was performed using Takara TB Green® Premix Ex TaqTM II (Takara, RR820). The primers are listed in Additional file 2: Table S3.
RNA-sequencing and analysis for differentially expressed transcripts
Total RNA was extracted using TRIzol (Invitrogen, Carlsbad, CA, USA). RNA sequencing analysis was technically supported by Aksomics Biotech CO. LTD (Shanghai, China). Differentially expressed genes were identified based on raw p values and fold change. Genes were considered significantly differentially expressed if the corrected p value was < 0.05 and the absolute fold change was > 2 in multiple tests.
Renal senescence-associated β-galactosidase (SA-β-gal) staining
Frozen Sects. (8 μm) were prepared for SA-β-Gal staining using the Senescence β-Galactosidase Staining Kit (Cell Signaling Technology, 9860).
Immunofluorescent (IF) staining
Paraffin-embedded kidney sections were deparaffinized and rehydrated. HK-2 cells cultured on coverslips were fixed with cold methanol/acetone for 10 min at room temperature. After blocking with 10% donkey serum for 60 min, the slides were immunostained overnight at 4 °C with primary antibodies: GLIS1 (Active Motif, 61,801), α-SMA (Proteintech, 67,735–1-Ig), COL I (Novus, NB600-408), FN (Abcam, ab2413), PPARα (Novus, NB300-537), ACOX1 (Abcam, ab184032), CPT1A (Proteintech, 15,184–1-AP), HK2 (Abcam, ab209847), PDK1 (Abcam, ab110025), METTL3 (Abcam, ab195352), and YTHDF1 (Abcam, ab252346), then incubated with a secondary antibody for 2 h. Cell nuclei were counterstained with DAPI (Sigma-Aldrich) for 10 min. Images were obtained using a confocal microscope.
Oil Red O staining
Oil Red O staining was performed on frozen kidney tissue Sects. (8 μm) and HK-2 cells cultured on coverslips (Sigma, O0625). The prepared samples were rinsed with distilled water and isopropanol, stained in Oil Red O working solution for 15 min, rinsed with 60% isopropanol and distilled water, and then counterstained with hematoxylin. Images were captured using a microscope.
Lactate and glucose measurement
Lactate concentration in kidney tissue and cell supernatant was measured using the Lactate Colorimetric Assay Kit (Biovision, K627-100) at a wavelength of 450 nm. Glucose concentration in the cell supernatant was detected using the Glucose Colorimetric/Fluorometric Assay Kit (Biovision, K606-100) at a wavelength of 450 nm.
Extracellular acidification rate (ECAR) measurement
The ECAR, representing the glycolysis ratio, was measured using a Seahorse Bioscience extracellular flux analyzer. Cells were cultured in XF24 V7 cell culture microplates (Seahorse Bioscience) with 10 mM glucose for 2 h at 37 °C, followed by the addition of the ATP synthase inhibitor oligomycin or the glycolysis inhibitor 2-deoxyglucose (2-DG). The results were analyzed using Wave software.
Assessment of cellular lipids
The levels of triglycerides (TG) (Cayman Chemical, 10,010,303), cholesterol (TC) (Cayman Chemical, 10,009,779), and free fatty acids (FFA) (Cayman Chemical, CAC-700310–96) in HK-2 cells were measured using commercial kits according to the manufacturer’s instructions.
Chromatin immunoprecipitation (ChIP) assay
ChIP was performed using the Chromatin Immunoprecipitation Kit (Millipore, 17–371). Fragmented chromatin from fixed cells was incubated with a GLIS1 antibody (Santa, Sc-365857) and protein G magnetic beads. DNA released from the precipitates was analyzed by PCR. The primer sequences specific to the GLIS1 binding region within the PPARα promoter region were as follows: PPARα promoter forward: 5′- TACGTGACCACTCTGCACAC-3′ and reverse: 5′- CCTCCGGGCTCAAAGACATT-3′. IgG and ddH2O served as negative controls, while RNA polymerase II and GAPDH were used as positive controls.
RNA immunoprecipitation (RIP) assay
RIP was conducted using the Magna RIP™ RNA-Binding Protein Immunoprecipitation Kit (Millipore, 17–701). Magnetic beads coated with 5 mg of specific antibodies against METTL3 (Proteintech, 10,573–1-AP), YTHDF1 (Proteintech, 17,479–1-AP), or rabbit IgG (Millipore) were incubated with cell lysates overnight. RNA–protein complexes were treated with proteinase K digestion buffer to isolate the immunoprecipitated RNA. The relative interaction between GLIS1 mRNA and METTL3/YTHDF1 was determined by PCR. The Primers for RIP-PCR were listed in Additional file 2: Table S4.
Methylated RNA immunoprecipitation (MeRIP)-qPCR
Poly(A) RNA was purified and fragmented using RNA fragmentation reagents (Thermo, AM8740). A fraction (1/10) of the RNA was reserved as the input control. Anti-m6A antibody (Abcam, ab151230) or mouse IgG was incubated with Protein A/G magnetic Beads, mixed with fragmented poly(A) RNA and 1 × poly(A) RNA RNase inhibitor-supplemented IP buffer, and rotated at 4 °C for 2 h. The m6A-containing RNAs were eluted and purified using an RNA purification column. RT-qPCR was then used to determine the enrichment of methylated mRNAs.
Luciferase reporter and mutation assay
GLIS1 mutants were generated using the Mut Express II Fast Mutagenesis Kit V2 (Vazyme). HK-2 cells were transfected with 250 ng of the 3′ UTR luciferase reporter plasmid (Promega). Luciferase activity was measured using the Dual Glo Luciferase Assay kit (Promega) as previously described [51].
RNA stability and protein decay assays
HK-2 cells, with or without siMETTL3 and siYTHDF1, were treated with 5 μg/mL actinomycin D (MedChemExpress, HY-17559). Total RNA was isolated at specified time points (0, 1, 2, 4, and 6 h), and the mRNA levels of GLIS1 were measured using RT-qPCR. To inhibit the proteasome, HK-2 cells, with or without siYTHDF1, were treated with 1.25 μM MG132 for 24 h, and GLIS1 protein levels were detected by western blot.
Ribosomal immunoprecipitation
The RPL22-Flag construct (Sino Biological, HG16861-NF) was transfected into HK-2 cells with vector or siYTHDF1. Protein lysates from vector control and siYTHDF1 cells were incubated overnight with 5 μg anti-FLAG antibody or IgG (control). RT-qPCR analysis was performed to determine the transcriptional changes of GLIS1.
Statistical analysis
All quantitative data were obtained from independent experiments with triplicate repeats and expressed as the mean ± SD. Statistical analysis was performed using SPSS 15.0 software, with two-tailed unpaired Student’s t-tests for comparisons between two groups and one-way ANOVA followed by Bonferroni test for multiple comparisons. A P value of less than 0.05 was considered statistically significant: *P < 0.05, ** P < 0.01, *** P < 0.001; NS indicates no significance.





留言 (0)