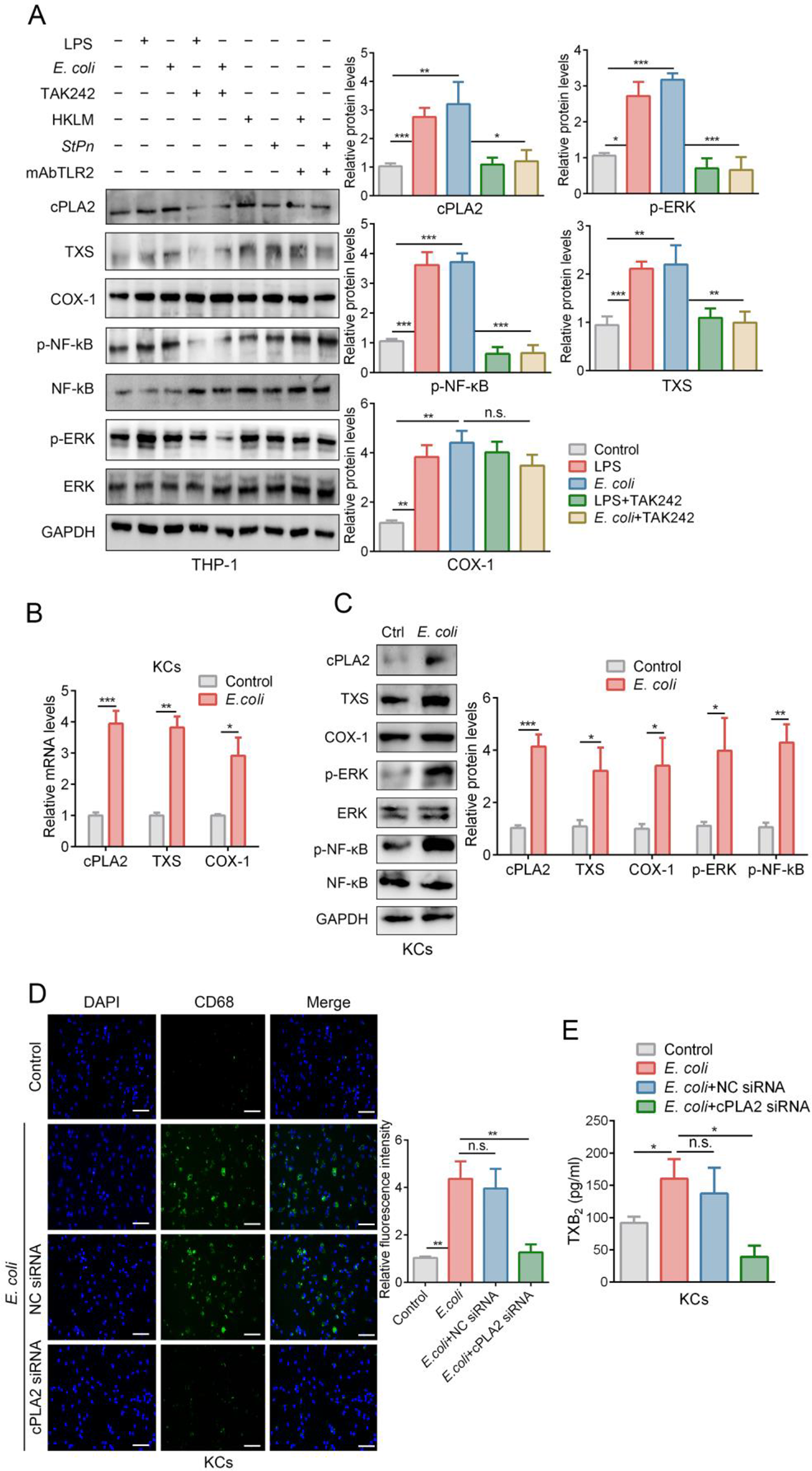
Cell culture and reagents
The human adrenocortical carcinoma cell line (H295R) was obtained from the American Type Culture Collection (CRL-2128; ATCC) and grown in H295R cell culture medium (CM-0399; Procell, Wuhan, China) at 37 °C in 5% CO2. 293T cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% FBS (Gibco, China). Cells were maintained in a humidified incubator at 37 °C with 5% CO2. Antibodies against TRIM2 (20356-1-AP, Proteintech), CYP11B2 (MABS1251, Millipore), CYP11B1 (MABS502, Millipore), β-Actin (sc-47778, Santa Cruz), MYC (18583, CST), GFP (ab290, Abcam), HA (TA180128, OriGene), and Ub (3933, CST) were purchased from specified commercial sources.
siRNA library screening
We procured the ON-TARGETplus siRNA library, which includes a pool of E3 ligase siRNAs, from Dharmacon (GE Healthcare, catalog G-105635-01). H295R cells were cultured in 96-well plates at a density of 4 × 103 cells per well. The transfection reagent (Lipofectamine 3000 Transfection Reagent) was diluted using Opti-MEM and then added to each well containing the siRNA pool. After a 15-minute incubation period, the siRNA (50 nM)/Opti-MEM/Lipo3000 mixture was transfected into the H295R cells. 48 h post-transfection, cells were harvested, and lysates were prepared for immunoblotting analysis to assess endogenous CYP11B2 expression. Detailed siRNA sequences for E3 ligases are provided in Table 1.
Table 1 The List of siRNA SequencesPlasmids and siRNAs
We subcloned HA-tagged TRIM2 (amino acids 1-744) and its truncations, including TRIM2-R (amino acids 1-111), TRIM2-BCC (amino acids 108–321), TRIM2-fn (amino acids 318–472), and TRIM2-NHL (amino acids 469–744), into the pcDNA 3.1 vector. Transfection of the plasmids into cells was performed using Lipofectamine 3000 transfection reagent (Thermo Fisher Scientific, USA) according to the manufacturer’s instructions, in serum-free OptiMEM medium (Gibco). The DNA construct sequences were thoroughly validated through DNA sequencing. For TRIM2 knockdown, the following siRNAs were utilized: TRIM2-si1: 5’-GGACGAUCUUAACCACCAA-3’, TRIM2-si2: 5’-GGGUGUAGCAGUGGAUUUCAA-3’, and siNC: 5’-UUCUCCGAACGGUGUCACGUTT-3’, all procured from Shanghai, China Gene Pharma Ltd. Opti-MEM medium, Lipofectamine® 3000, and P3000TM (Invitrogen) were employed for transfecting cells with siRNA and plasmids according to manufacturer’s instructions after the H295R cells reached 80–90% confluent. The transfection efficiency was detected by quantitative polymerase chain reaction (PCR) at 24 h after transfection.
Measurement of aldosterone concentration
When H295R cells were grown to confluence in 24-multiwell plates, they were transfected with HA-TRIM2 or empty vector for 48 h. Angiotensin II (ANGII) was then added to the media at a concentration of 100 nmol/L, and the cells were incubated for 6 h. Before treating the cells with ANGII, they were incubated in serum-free medium for 24 h. Aldosterone concentrations of the media were thereafter measured by Aldosterone EIA kit (Cayman Chemical, Ann Arbor, MI, USA) according to the manufacturer’s instructions.
Total RNA isolation and qRT-PCR
We isolated total RNA from APA cells using the RNeasy Mini Kit (Cat. #74101, Qiagen, Germany) according to the provided protocol. Subsequently, cDNA synthesis was performed using the ReverTrace qRT-PCR Kit (Toyobo, China) for reverse transcription reactions. Each qRT-PCR reaction consisted of 3 μl primer, 4.5 μl cDNA, and 7.5 μl iQTM SYBR® Green Supermix (Bio-Rad, USA). The primer sequences for qRT-PCR are provided in Table 2. The cycle threshold (Ct) was determined relative to GAPDH.
Table 2 The specific primer sequencesProliferation assays
The MTT test involved seeding 3000 BLCA cells per well in a 96-well plate. Next, 20 μL of MTT solution was added to every well, and the plate was subjected to a 4-hour incubation period. A microplate reader (SpectraMax M2, USA) was used to measure the absorption values at 540 nm after the precipitate was dissolved in DMSO. A colony formation assay was performed for 10 days on a 6-well dish containing 1000 cells per well. Then, 4% paraformaldehyde was used to fix the colonies, and 0.1% crystal violet was used to stain them.
Western blotting
The process of total protein isolation and western blotting commenced with the centrifugation and ice-based lysis of cells for a duration of thirty minutes, employing RIPA buffer supplemented with protease inhibitor and phosphatase inhibitor (Sigma-Aldrich, USA). Subsequently, the cells underwent treatment using an ultrasonic crusher for 10 s followed by centrifugation at 14,000 × g for 10 min. Protein separation and detection procedures were executed in accordance with the methods previously outlined [28, 29]. Finally, immune response bands were visualized using the electromagnetic interference XRS imaging system (Bio-Rad, USA).
Co-immunoprecipitation
Following transfection for 48 h, cells were harvested and subjected to lysis using a cell lysis buffer containing a proteasome inhibitor. The lysis process was conducted on ice for 30 min, followed by centrifugation at 14,000 × g at 4 °C for 10 min. A small aliquot of the resulting supernatant was utilized as Input for western blot analysis. Magnetic beads, bound to the corresponding antibodies, were then added to the remaining supernatant and allowed to incubate at 4 °C with gentle shaking overnight to facilitate the conjugation of antigen-antibody magnetic beads. After the immunoprecipitation reaction, the magnetic bead coupling complex was washed with buffer. Following this, 15 μl of 1 × SDS buffer was added and the mixture was heated at 100 °C for 5 min. The proteins were then separated via SDS-PAGE electrophoresis, followed by western blotting to identify the interacting proteins.
Immunofluorescence
H295R cells underwent a series of procedures: they were initially washed with PBS and subsequently fixed with 4% PFA for 20 min at room temperature. Following fixation, the cells were again washed three times with PBS and then subjected to overnight incubation at 4 °C with the desired antibodies. After another three washes with PBS, the cells were incubated with fluorescein-labeled secondary antibodies for 40 min at 37 °C. Following an additional three washes with PBS, the cells were incubated with DAPI (D8417, Sigma-Aldrich, Germany) at room temperature for 30 min and mounted in mounting medium. The slides were subsequently analyzed using a laser scanning confocal microscope (LSM880, Zeiss, Germany).
Human adrenal tissues and immunohistochemistry
The human adrenal tissue samples used in this study were obtained from primary aldosteronism (PA) patients who underwent lateral adrenalectomy at Zhongnan Hospital of Wuhan University. The diagnosis of PA was determined according to the clinical practice guidelines of the American Endocrine Society [30]. Normal adrenal tissues were obtained from the adjacent tissue within 1 cm of the tumor margins. All adrenal tumors and normal adjacent tissues were pathologically confirmed. Patients provided informed consent for participation in the study. The protein expressions of CYP11B2 and TRIM2 in human aldosterone-producing adenomas (APAs) and normal adrenal tissues were detected using immunohistochemistry (IHC). Initially, fresh tumors were fixed in 4% paraformaldehyde (PFA) for 24 h. Subsequently, they were embedded in paraffin and sectioned into 5 μm slices. The sections were then subjected to IHC staining using the CYP11B2 antibody (MABS1251, Millipore, dilution 1:200) and the TRIM2 antibody (20356-1-AP, Proteintech, dilution 1:200). The DAB chromogen was applied for incubation, followed by counterstaining with hematoxylin. Finally, the IHC-detected images were scanned and analyzed using a molecular microscope (Olympus BX53). In this study, tissue microarrays (TMAs) and IHC analysis were also utilized to assess protein expression and distribution, which have been widely applied in previous studies [28]. The IHC staining scores were evaluated using the following criteria: 0 indicated negative staining, 1 indicated weak positive staining, 2 indicated moderate positive staining, and 3 indicated strong positive staining. The positivity rate was assessed based on the percentage of cells showing positive staining, with the scoring as follows: 0 indicated negative staining, 1–25% scored 1, 26–50% scored 2, 51–75% scored 3, and 76–100% scored 4. To calculate the total staining score, the staining intensity score was multiplied by the staining positivity rate score. The immunohistochemical grading was performed in a blinded manner by two independent pathologists.
Mass spectrometry analysis
After transfecting plasmids into H295R cells, the cells were incubated for 48 h and IP was performed. After the immunoprecipitation reaction, the magnetic bead coupling complex was washed with cold PBS buffer. Mass spectrometry analysis was conducted using timsTOF Pro (Bruker, USA). The mass spectrometry data were processed using MaxQuant (V1.6.6) software and the Andromeda database search algorithm. The Proteome Reference Database for Humans in UniProt was used for the search.
Molecular docking
Molecular docking is utilized to validate the binding activity between proteins or between proteins and small molecules. The HDOCK online platform (http://hdock.phys.hust.edu.cn/) was employed for molecular docking in this study. HDOCK can analyze different conformations of protein-protein docking, binding activities under different conformations, and amino acid residues within 5Å of interaction distance. Proteins with UniProt IDs P19099 and Q9C040, corresponding to CYP11B2 and TRIM2, were downloaded from the UniProt database and subjected to docking analysis. Ligplus software can be used to analyze the two-dimensional interactions between the two proteins. PyMOL (version 4.3.0) software is employed for visualizing the amino acid residues involved in the interaction between the two proteins.
Statistical analyses
The data are presented as the mean ± standard deviation (SD) of three independent experiments. The two-tailed t-test was used to analyze normally distributed data, while non-parametric Kruskal-Wallis test was used for non-normally distributed data. Data with more than two groups using one-way ANOVA. Spearman’s test was employed to investigate the correlations between variables. All statistical analyses were performed using GraphPad Prism 9.00 software. Image analysis and quantification were conducted using ImageJ v1.45 software. In statistical analysis, p-values less than 0.05 were considered statistically significant.








留言 (0)