
記住我


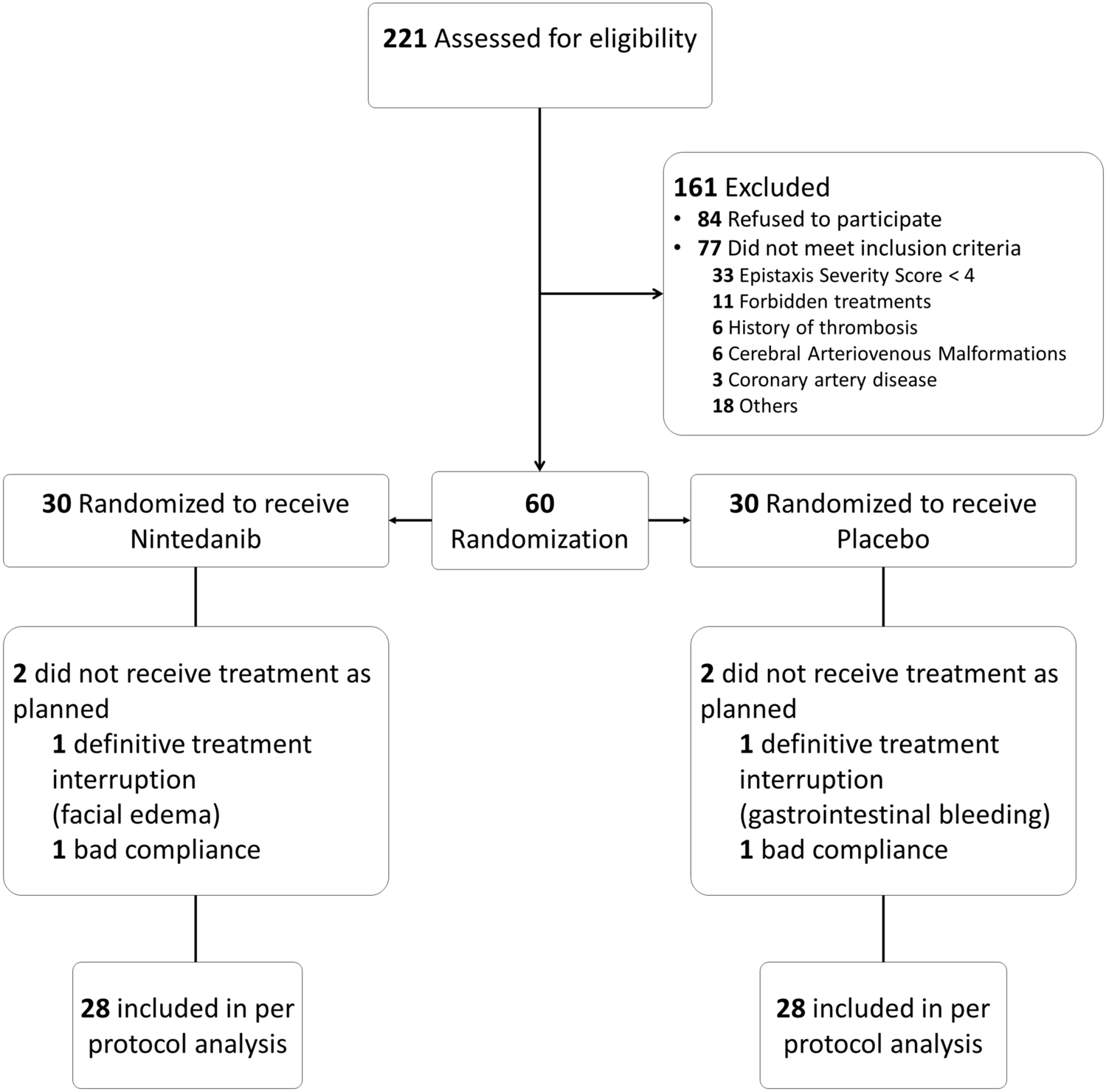






This phase II study was a national, prospective, multicenter trial, comparing nintedanib to placebo in a parallel-group, randomized design with a 1:1 ratio. It was conducted in a double-blind setting, with both the treatment duration and follow-up period set at 12 weeks.
It was approved by the research ethics committee and authorized by the French medical products agency (ANSM). All patients gave their oral and written consent in accordance with national regulations. The study was registered in a public trials registry (clinicaltrials.gov identifier NCT03954782).
ParticipantsThis study enrolled patients aged 18 years and older, with clinically confirmed HHT and moderate to severe epistaxis defined by an epistaxis severity score (ESS) greater than 4.
Patients in the HHT network were informed during a standard follow-up consultation in an HHT center. All French HHT centers were involved in patient selection, but treatments were centralized in ten hospitals across the country.
Exclusion criteria included the presence of non-treated pulmonary AVMs, hemoptysis, hematuria, overt gastro-intestinal bleeding or ulcers within 12 months, cerebral AVM on MRI, having a liver disease or renal failure or active infection, known coronary artery disease or predisposition to thrombosis, use of certain medications (e.g., anticoagulant or antiplatelet therapies, other anti-angiogenic treatments, P-glycoprotein substrates/inducers/inhibitor treatments), presence of unhealed wounds or recent surgery; having QTc prolongation.
Randomization and blindingThe randomization process was centralized. A unique list for all centers was generated using SAS® by the Pôle de Santé Publique at the Hospices Civils de Lyon—clinical research unit, using random block sizes of 4 and 6. Patients included were randomly assigned to one of two treatment groups using the IWRS (interactive web response system) based on this list. The software ENNOV clinical version 7.1 (clinsight) was used for the data-management.
InterventionsIn the nintedanib group, patients received 150 mg of nintedanib twice daily, administered orally approximately 12 h apart every day for 12 weeks. The nintedanib was manufactured by the company Boehringer Ingelheim and commercialized as OFEV® 100 mg and 150 mg containing respectively 100 mg and 150 mg of nintedanib as esilate. The comparative treatment was a placebo provided by the same company as soft gelatin capsules (100 mg and 150 mg) containing a suspension of titanium dioxide as the drug substance substitute, and identical in appearance to the nintedanib. If a dose was missed, administration resumed at the next scheduled time at the recommended dose and no additional dose was taken so as not to exceed the recommended maximum daily dose of 300 mg. In case of adverse reactions, the treatment could be temporarily interrupted or adjusted to 200 mg (100 mg twice daily). If adverse reactions persisted after the dose reduction, the treatment was discontinued.
OutcomesPrimary outcomeThe primary endpoint was the proportion of patients achieving a reduction of at least 50% in mean monthly duration of epistaxis in the last 8 weeks of treatment (P2) as compared to the 8 weeks before treatment (P1). This criterion was assessed by monitoring epistaxis grids filled in daily by the patients and collected at each visit or filled in online. These grids contained the number of episodes per day and their duration. The mean monthly duration of epistaxis on each reporting period was computed over the last 56 days or less (corresponding to 8 weeks) and normalized/reduced to 28 days, considering that 1 month is equal to 4 weeks, thanks to the following formula: \(28* \frac_^epistaxis\,daily\,duration}\)
Secondary outcomesEpistaxis was also assessed with regard to.
Epistaxis monthly duration as a continuous variable and epistaxis frequency before, during and after treatment.
Reduction of at least 50% in the mean monthly duration of epistaxis during the last 8 weeks of follow-up (P3) as compared to the 8 weeks before treatment (P1).
Epistaxis severity score (ESS) at the inclusion visit, end of treatment, and end of follow-up.
Other clinical criteria were also assessed, such as quality of life using SF-36 questionnaires (filled out by the patients at the inclusion visit, at the end of the treatment and follow-up periods), and number of red blood cell (RBC) units transfused and iron infusions (for the 8 weeks before treatment, during the last 8 weeks of treatment and follow-up periods). National recommendations were used regarding indications for RBC unit transfusions. No specific protocol was implemented for iron supplementation during the study.
Biological criteria such as hemoglobin and ferritin levels were also measured at inclusion, and at the end of the treatment and follow-up periods. The different criteria were assessed during the six on site visits.
Safety criteria: all adverse events (AE) and severe AE were collected throughout the study and coded using the medical dictionary for regulatory activities (MEDDRA) and graded according to the common terminology criteria for AE (CTCAE) classification. A safety committee reviewed all the adverse events collected and their relationship to the study treatment.
Sample size calculationWe hypothesized that 60% of patients would show improvement in the treatment group against 15% in the placebo group. Twenty-seven patients included in each group would make it possible to attain a power of 90% according to a Fisher’s exact test, leading to 54 patients overall.
Considering early withdrawal and patients who may be lost to follow-up, we decided to include 30 patients in each group, leading to a total of 60 patients.
Statistical methodsBaseline characteristics were summarized as number of patients (%) for categorical variables and as median (Q1–Q3) and mean (SD) for continuous variables. All analyses were carried out on the ITT population, only the main analysis on the primary endpoint was also performed on the PP population.
Between-group differences were tested using the Mann–Whitney test for quantitative outcomes and the Fisher test for qualitative outcomes (both non-parametric tests). A two-sided p-value of less than 0.05 was considered statistically significant. No adjustment for multiple testing was performed. Two-sided 95% confidence intervals were used.
For patients with fewer than 14 days (inclusive) missing on epistaxis grids, the mean monthly duration was computed on the data observed (from the 8-week, 56-day period evaluated). For patients with more than 14 days missing on the grids in P3, the result for categorical data was imputed as a failure in the nintedanib group and success in the placebo group.
All statistical analyses were performed using R software version 4.1.1.
留言 (0)