Trial design and oversight
OPTIFLUID is a multicenter, single-blind, randomized, parallel, controlled, pilot trial. Patients were recruited between September 20, 2021, and February 25, 2023 in ICUs in the university hospitals of Nîmes and Dijon, and the general hospital of Alès, France. The study was approved by the Comité de Protection des Personnes Sud-Méditerranée II, France (number 2020-A01952-37). Written informed consent was obtained from patients or their legal surrogates either before randomization or as soon as possible thereafter [14]. The trial was funded by Nîmes University Hospital, France.
Patients
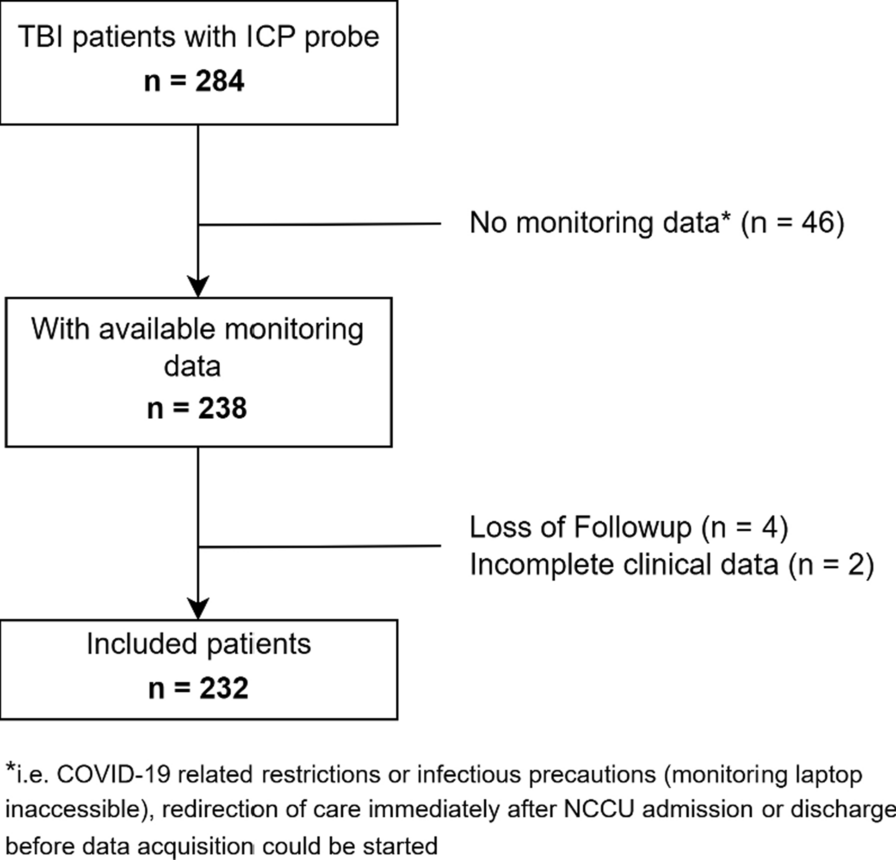
Patients between 18 and 85 years’ old were eligible if they were admitted to the ICU in the early phase of septic shock (within 24 h after the start of vasopressor therapy). Septic shock was defined as suspected or confirmed infection, a plasma lactate level of ≥ 2 mmol/L and ongoing infusion of a vasopressor agent [1]. Exclusion criteria were acute kidney injury requiring renal replacement therapy within the next 24 h, end stage chronic kidney disease, and severe malnutrition with body mass index < 18. Detailed inclusion and exclusion criteria are presented in Supplementary Table S1.
Randomization and blinding
Randomization was performed using a centralized, computer-generated allocation sequence. Eligible patients were randomly assigned in a 1:1 ratio, (in permuted blocks of 2), to receive the optimized restrictive strategy or standard fluid therapy (control group).
Treatment group assignments were not masked for clinicians or investigators but were concealed from patients and the statisticians.
Interventions
All patients were treated according to the recommendations of the Surviving Sepsis Campaign [15]. During the first 7 days of septic shock, fluid infusion was given following an optimized restrictive strategy or standard fluid therapy (control group). To calculate the dose of fluids administered, we always used the patient's weight on admission to ICU. Trial staff (physicians, clinical pharmacists and nurses) were trained before inclusion began. The coordinating investigator and the clinical pharmacist were available throughout the inclusion period to help the investigators comply with the protocol.
Optimized restrictive strategy (experimental group)
A restrictive fluid protocol was applied upon inclusion on Day 1 and continued for the first 7 days of hospitalization in the ICU. Clinicians were instructed to reduce fluid intake as much as possible in terms of maintenance fluids, drug dilution and artificial nutrition. The management of resuscitative fluids, defined as fluids prescribed to optimize blood volume in the treatment of shock, was not modified by the protocol, and was left to the judgement of clinicians according to their usual monitoring strategy (cardiac ultrasound, pulse index continuous cardiac output, right heart catheterization, central venous pressure, etc.). For drug dilutions, the clinicians consulted charts (Supplementary Table S2) showing the maximum possible concentrations for the drugs most frequently used in ICU, particularly antibiotics, based on literature data [16]. For artificial nutrition, the intravenous route was discouraged before Day 5 [17]. For enteral nutrition, the protocol provided for the use of a hypercaloric (2 kcal/ml) high protein product, with a total caloric objective of 20 kcal/kg per day in the acute phase and 30 kcal/kg per day after stabilization [18]. Intravenous fluids other than those needed to dilute drugs and electrolytes were prohibited, and only 2 ml/hour of saline or glucose were authorized to maintain veins. A clinical pharmacist intervened daily to check adherence to the fluid restriction protocol. If the restrictive strategy induced hypovolemia and/or biological abnormalities (sodium, potassium, chloride, etc.), the treating physician could increase fluid therapy in accordance with the protocol's recommendations. The fluid restriction strategy did not involve the systematic administration of diuretics.
Standard fluid strategy (control group)
The choice of the volume of fluid intake other than resuscitative fluids was left to the physician’s discretion, based on usual practices: usually between 500 and 2000 ml/day of maintenance volumes. The dilutions of drugs were those officially planned at the time of marketing approval, enteral artificial nutrition (isocaloric, high protein) was initiated as soon as possible, with the same rules for parenteral nutrition if the caloric target could not be reached with enteral nutrition.
Diuretics were authorized in both groups at the judgement of the clinicians according to the needs of the patients, but were not recommended as long as patients had high dose vasopressors.
Outcomes
The primary outcome was fluid balance in the first 5 days. Secondary outcomes were fluid balance at Day 3, 5 and 7; body weight variation at Day 5 and 7; death from any cause at Day 7 and 28; the number of days alive without life support and without organ failure (SOFA = 0) at Day 28; the number of days alive without vasopressors, mechanical ventilation and renal replacement therapy at Day 28; the number of days alive outside the ICU at Day 28; the ICU and hospital length of stay; and the use of diuretics (cumulative dose administered over 7 days). Finally, in addition to unexpected adverse events, predefined adverse events during the entire ICU stay were collected, specifically metabolic (mild and severe hypernatremia, hyponatremia), renal (hyperkalemia, Acute Kidney Injury KDIGO2 and 3), and nutritional (hypoglycemia, stage 3 pressure sores acquired in the ICU); cumulative insulin dose over 7 days) complications potentially related to the restrictive fluid strategy; norepinephrine doses and lactate levels over 7 days.
Statistical analysis
Considering that "hidden" fluid intake constitutes 55% of total intake by patients, we estimated that a restrictive strategy would lead to a relative 40% reduction in fluid balance, conferring a significant clinical impact. Assuming a mean fluid balance of 70 mL/kg in the control group [6,7,8], 40 mL/kg in the experimental group and a standard deviation of 35 mL/kg, we estimated that 44 patients (22 per group) were needed to guarantee 80% statistical power with a two-sided alpha risk of 5%. This number was increased by 10% to take into account any deaths between Day 1 and Day 5. The total number of subjects required was therefore 50 patients (25 per group).
The primary endpoint, fluid balance at Day 5 (ml/kg), was compared between groups using a Mann–Whitney test via intention-to-treat analysis. A second per-protocol analysis was performed, where only patients who had been depleted for at least 5 days were considered, as well as a sensitivity analysis in which we excluded patients with bleeding (defined as more than 2 units of blood transfusion in the same day). Categorical variables are expressed as numbers and percentages; continuous variables are expressed as means and standard deviations or medians and interquartile ranges (IQR) as appropriate. Quantitative variables were compared between two groups using a Student's t test if the variable had a normal distribution, or a Wilcoxon Mann Whitney otherwise. The mean difference between the two groups was also calculated with the 95% confidence interval. For categorical variables, either the Chi2 test or Fisher's exact test were used and the percentage difference between the experimental and control group was also calculated, as was the associated relative risk with the 95% confidence interval. The length of ICU and hospital stay, and the number of days free of mechanical ventilation, vasopressors, and renal-replacement therapy are expressed as medians and IQR and were compared using the Mann–Whitney test. We evaluated safety by calculating the percentage of patients in the two groups presenting metabolic, renal and nutritional adverse events; percentages were compared with the use of appropriate tests. The significance level was set at 0.05 for all analyses.
Analyses were performed with SAS Enterprise Guide software, version 7.1.
Study protocol
The trial protocol, which includes the statistical analysis plan, was registered on ClinicalTrials.gov with the identifier NCT04947904.
For evaluation of patient safety, this trial has been registered with France National Agency of Drug Safety (Agence Nationale de Sécurité du Médicament et des Produits de Santé, ANSM).
Trial data were monitored at the sites by independent monitors and monitored centrally by staff at the coordinating centre. The first and the last author wrote the first draft of the manuscript, which was reviewed by all the authors. Statistical analyses were performed by the trial statistician in accordance with International Conference on Harmonisation Good Clinical Practice guidelines. The authors vouch for the accuracy and completeness of the reported analyses and for the fidelity of the trial to the protocol.


留言 (0)