
記住我
Colorectal cancer (CRC) is a heterogeneous disease with different underlying mechanisms leading to different molecular subtypes with varying treatment responses (1). Around 15% of sporadic CRCs are classified as microsatellite instability-high (MSI-high) or DNA mismatch repair-deficient (dMMR); these tumors accumulate a higher burden of somatic mutations, high antigen burden, and high infiltration of T cell lymphocytes overall (2, 3). Given their characteristic high infiltration of T cells, MSI-high tumors are more likely to respond to immune checkpoint inhibitors (ICIs), which has resulted in substantial, durable improvement in treatment of MSI-high CRC (4–6). MSI-high status is strongly correlated with hypermutation status, where around 16% of CRCs are hypermutated, three quarters of which are MSI-high (7, 8). Hypermutation can be caused by multiple factors, including MMR deficiency related to MSI as well as POLE mutations that can lead to a hypermutated phenotype without MSI (9).
Although previous studies have shown MSI-high status to be associated with T cell infiltration in CRC (10–13), specifically CD45RO+ T cells (14), characterization of infiltrating T cells in these studies has been limited. Specifically, many studies have measured T cell densities using single-plex immunohistochemistry assays to assess individual cell markers, which fails to represent the complexity of T cell response and diverse T cell subsets. A more granular understanding of specific T cell subsets that are associated with the MSI pathway, a strong determinant of prognosis and treatment, may help inform targeted immunotherapy treatment decision-making for both the MSI-high and MSI-low/MSS (microsatellite stable) CRC subtypes.
Utilizing the resources of studies participating in the Genetics and Epidemiology of Colorectal Cancer Consortium (GECCO), we aimed to characterize T-cell response in diverse subsets of CRC tumors using a multiplex immunofluorescence panel and identify which specific T cell subsets, in either epithelial or stromal tumor area regions, are associated with MSI-high and hypermutation status.
MethodsStudy populationThis study was conducted within a subset of studies participating in the Genetics and Epidemiology of Colorectal Cancer Consortium (GECCO) for which tumor immune profiling data were available, including: the Ontario Family Colon Cancer Registry (OFCCR) (15), the Nurses’ Health Study (NHS) (16), and the Health Professionals Follow-up Study (HPFS) (16). The NHS and HPFS cohorts were the basis of the Prospective Cohort Incident Tumor Biobank (PCITB) (17, 18). Data on tumor tissue analyses and other associated metadata constituted the entirety of the PCITB (17, 18). After excluding participants with tumor immune profiling data who did not have data on MSI status available (N=79), data from a total of N=1,236 individuals was available for analysis. The GECCO consortium is an international collaboration that focuses on the identification and characterization of genetic risk factors and gene-environment interactions for CRC and investigates tumor genome/characteristics, microbiome, and immune response (19, 20). Clinical and epidemiologic data were collected by each study through self-reported structured questionnaires or in-person interviews. All participants gave written informed consent and studies were approved by their respective Institutional Review Boards. Studies identified incident CRC either via self-report of diagnosis from study participants, with confirmation via adjudication of medical records (NHS, HPFS), or via population-based cancer registries, regional hospitals, or healthcare management organizations (OFCCR).
Microsatellite instability (MSI) and hypermutation status callingMicrosatellite instability (MSI) status information was collected by each study according to individual study protocols. The harmonization procedures as well as the methods for individual studies have been previously described (15, 21–26). NHS and HPFS (24) used polymerase chain reaction (PCR) based assessment of MSI status, while OFCCR (15, 25) utilized a combination of PCR-based and immunohistochemical (IHC) assays for MSI. For all studies, tumors were classified as MSI-high if 30% or more of the markers showed instability and non MSI-high if < 30% and > 0% showed instability (MSI-low), or if no marker exhibited instability (microsatellite stable, MSS). To harmonize markers across all studies, we created two categories for downstream analyses, MSI-high and non MSI-high (MSI-low/MSS).
Previously collected targeted sequencing data was available for a subset of participants within the present study to quantify hypermutation status (N=639). Briefly, tumors from OFCCR were sequenced with a 1.34 megabase (Mb) targeted panel covering 205 genes (27), and tumors from NHS and HPFS were sequenced with a 1.96Mb targeted panel expanding the panel to 298 genes. DNA was extracted from formalin-fixed paraffin-embedded (FFPE) CRC tissue that was macrodissected from tissue slides. Matching normal DNA was primarily extracted from either adjacent normal colonic FFPE tissue or peripheral blood. Hypermutation status was defined by plotting point mutations for all samples within each targeted sequencing batch, where two distinct peaks were observed. The minimum value between peaks was used as a cut-point in each sequenced dataset, which were 23 and 26 point mutations, respectively (27).
T cell profilingWe profiled the in-situ T cell landscape of CRC using a multiplexed immunofluorescence (mIF) panel. Tissue microarray (TMA) construction using standard methods has been described in detail elsewhere (28). In brief, core selection from formalin-fixed paraffin-embedded (FFPE) blocks was guided by pathologist review of H&E-stained slides. For the vast majority of CRC cases, 2-4 cores (approximately 0.6 mm) from tumor areas were placed into a TMA block. For a small number of participants only a single core was available due to tissue detachment before and/or after the staining process. TMAs were mounted on negatively charged slides, which generally provide good tissue section adherence. T cell immune microenvironment was assessed for each tumor histologically using a multispectral imaging platform (PhenoImager HT, Akoya Biosciences, Marlborough, MA, USA) for mIF, where TMAs were stained with all markers concurrently, imaged, segmented into epithelial and stromal regions as well as individual cells, phenotyped, and finally quantified into counts. TMAs were magnified at 0.5 um/pixel through the PhenoImager HT platform. Assays were conducted on TMAs using a customized 9-plex panel that included antibodies targeting CD3, CD4, CD8, PTPRC (CD45RO, CD45RA), FOXP3, MKI67 (Ki-67; proliferation), as well as a KRT (keratin, pan-cytokeratin) antibody to identify tumor cells and nuclear DAPI (di-(4-amidinophenyl)-1H-indole-6-carboxamidine) stain. We used the Opal multiplex technique that is a well-established method for detecting multiple biomarkers in a single tissue section (29). To assess potential signal interference for each marker, we stained human lymph node tissue and tonsil tissue without cancer metastasis as a control and set up the fluorescence unmixing condition. Additionally, to minimize background fluorescence and signal interference, multiplex immunofluorescence analysis was performed on autofluorescence slides treated with Opal fluorescences, excluding primary antibodies. The antibodies and staining conditions for multiplex immunofluorescence histological analysis are presented in Supplementary Table 1. This marker panel was validated against traditional chromogenic immunohistochemistry, where we performed single-plex immunofluorescence for each marker to assess the performance of T-cell-targeted multiplex immunofluorescence. This involved comparing the single immunohistochemical staining of each marker with lymphoid tissue as a reference.
With pathologist supervision, 9-plex digital fluorescence images were processed using supervised machine learning (inForm 2.6.0, Akoya Biosciences, Marlborough, Massachusetts, U.S.) to segment each region of interest into epithelial and stromal tissue areas. The concomitant single-cell-level analysis was performed using R v.4.3.0. (R Foundation for Statistical Computing, Vienna, Austria). T cell densities (cell count per mm2) were then quantified within each TMA core, where we were able to identify naïve, memory, and regulatory helper T cells, naïve, memory, and regulatory cytotoxic T cells, double-negative naïve and memory T cells, and CD3- immune cells based on marker co-localization, all within either epithelial or stromal tissue, resulting in 18 unique subsets (Table 1).
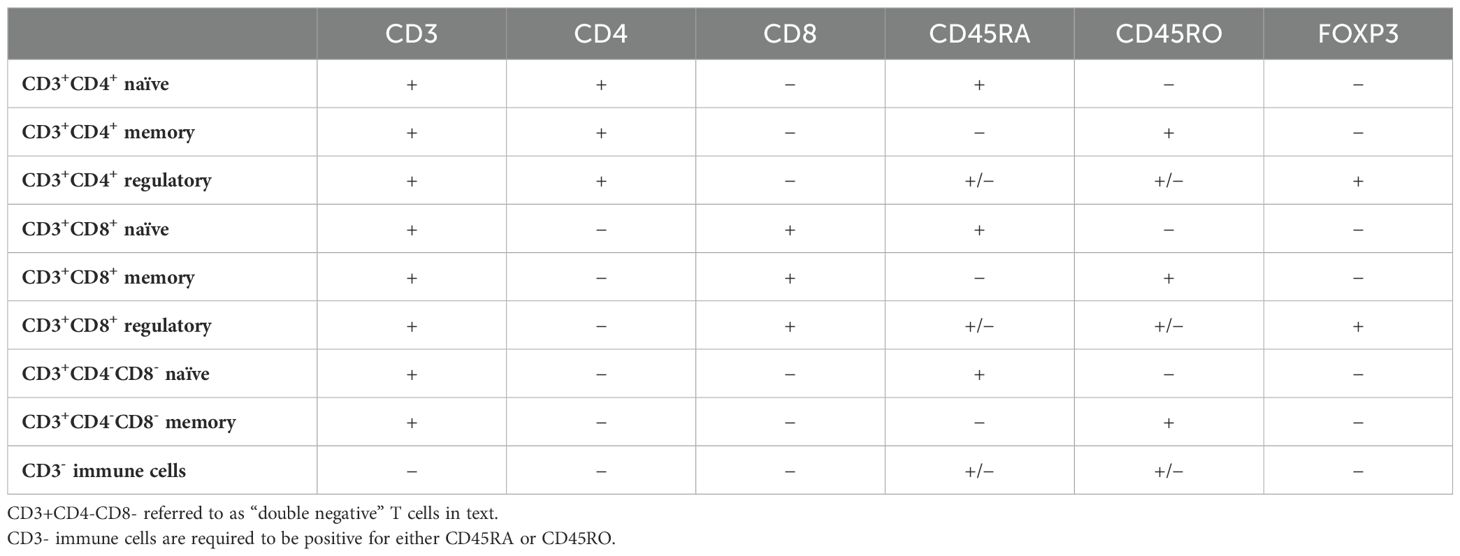
Table 1. T cell subset definitions by marker co-expression.
Statistical analysisFor each CRC case, we used the density metrics of each T cell subset in tumor epithelial or stromal tissue areas averaged across multiple TMA cores. Box plots, including median and interquartile ranges, were used to visualize distributions of T cell subset densities. Wilcoxon non-parametric tests were used to assess for significant differences between MSI-high and MSI-low/MSS for each T cell subset. Participant characteristics were described for the total study population and by MSI status using mean and standard deviation for continuous age, or frequencies and percentages for categorical variables. P values were calculated using t-tests for mean differences for age or chi square for frequencies for categorical variables. Scatter plots and Spearman correlation coefficients were used to determine the association between continuous tumor mutational burden (indels and single nucleotide variants) and T cell densities.
We used multivariable ordinal logistic regression or multivariable binary logistic regression to estimate odds ratios (ORs) and 95% confidence intervals (CIs) for the association of MSI or hypermutation status with quantiles of specific T cell densities in CRC, depending on the percentage of zeros within each subset. Ordinal logistic regression either used quartiles or tertiles of T-cell densities as the outcome, where different subsets were assessed for zero-inflation considering both epithelial and stromal tissue, so the same subset used the same categorization in each tissue area (Table 2). To further investigate the impact of MSI status on T cell subsets, we stratified our non-overlapping immune cell subsets by proliferation (MK167, Ki-67) status. When categorized additionally by proliferation status, the same categorization was used for each subset in both proliferating and non-proliferating subsets, as well as in both epithelial and stromal tissue areas (Table 3). Subsets with less than 25% zero densities were divided into quartiles, subsets with greater than 25% and less than 60% zero densities into tertiles, and greater than 60% zeros were categorized as binary, and therefore used binary logistic regression. Study batch-specific quantile cut points were used to limit potential batch effects across TMAs. Models were adjusted for age, sex, study batch (NHS & HPFS, OFCCR), and cancer site (proximal, distal, rectal, missing). P-values were adjusted for multiple testing using the false discovery rate (FDR) method for 18 independent tests in primary analyses and 36 independent tests when further stratified by proliferation status, where FDR adjusted P values less than 0.05 were considered significant (30). All P values reported are two-sided. All analyses were performed using R, version 4.4.0 (R Foundation for Statistical Computing, Vienna, Austria) software.
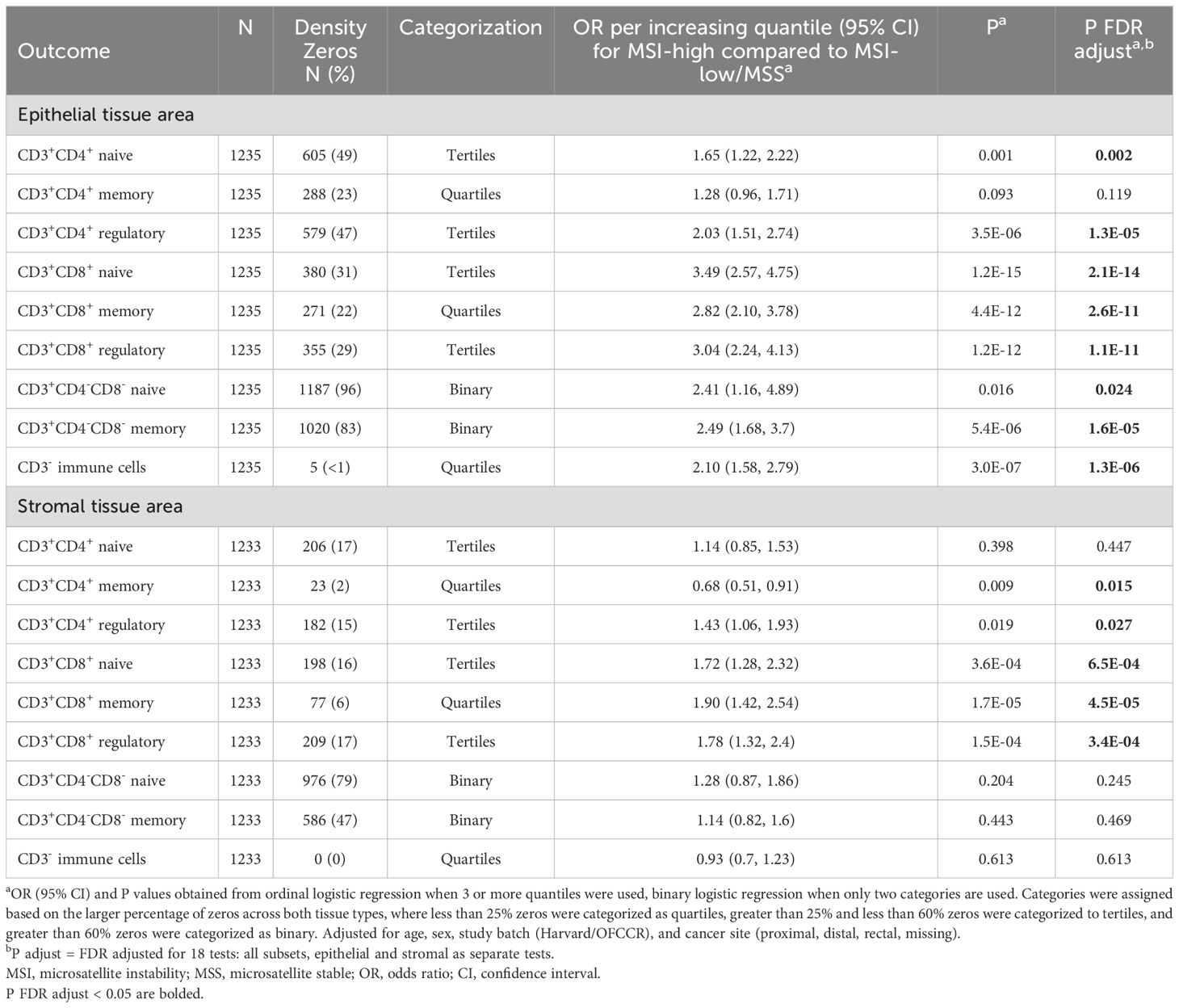
Table 2. Association between microsatellite instability status and T cell subset densities.
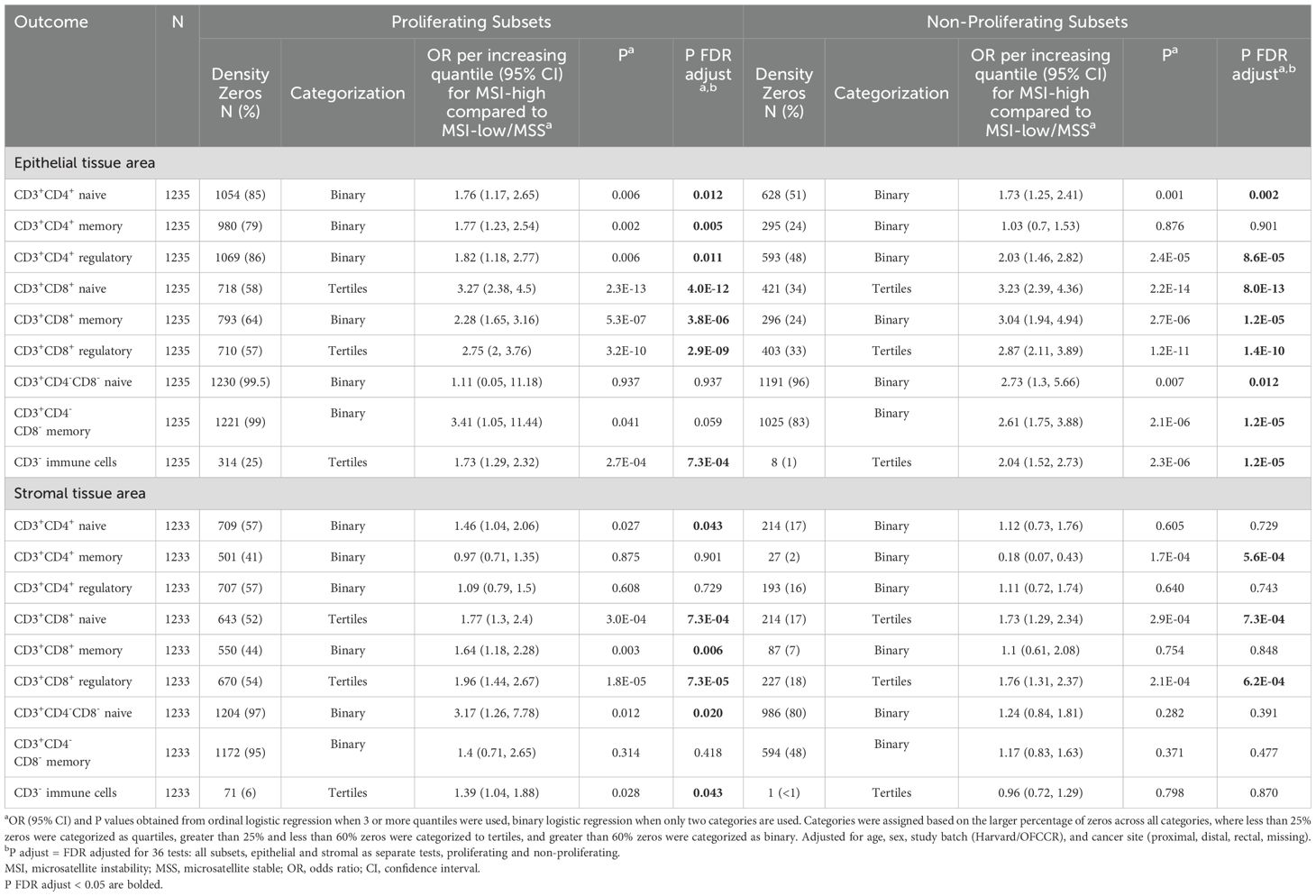
Table 3. Association between microsatellite instability status and T cell subset densities, stratified by proliferation (MKI67) status.
ResultsParticipant cohort and experimental approachThe mean age at diagnosis (standard deviation) of participants was 67 (9) years (Supplementary Table 2). Proportions of both sexes were roughly equal, where 54% of our sample were female. A majority of included participants had tumors located in the colon (77%), and were diagnosed at stages II or III (60%). In total, 218 out of 1,236 tumors were classified as MSI-high (18%), and among those with targeted sequencing data, 107 (17%) were hypermutated. Compared to MSI-low/MSS cases, MSI-high cases were more likely to be female, belong to the NHS study, have tumors located in the proximal colon, and be diagnosed at stage II or III (Supplementary Table 2).
In this study we identified 2,627,801 total immune cells, including 572,163 CD3+ T cells. One participant did not have epithelial tissue regions and three participants did not have any stromal tissue regions present in their TMA core, resulting in 1,235 participants for tests of epithelial tissue area and 1,233 for tests of stromal tissue area. A schematic outlining our T cell populations identified, example epithelial tissue areas and stromal tissue areas, and example cell segmentation and multiplex images are presented in Supplementary Figure 1. T cell subsets were overdispersed and right-skewed (Supplementary Figure 2). Stromal tissue regions generally had higher cell densities than epithelial regions for each T cell subset. The percentage of zeros for each T cell subset was generally higher in epithelial than stromal tissue regions (Table 2). Double-negative (CD3+CD4-CD8-) naïve and memory T cells in epithelial areas were the rarest subsets, resulting in the most zeros (96% and 83% zeros, respectively). CD3+CD4+ and CD3+CD8+ T cells had more modest zero percentages, ranging from 2% to 49%.
T cell density differences by MSI statusRepresentative multiplex immunofluorescence images of an MSI-high tumor and MSI-low/MSS tumor are shown in Figure 1. MSI-high tumors had significantly (Wilcoxon pFDR adjusted < 0.05) higher densities of all CD3+CD4+ and CD3+CD8+ T cells in epithelial tissue areas, as well as higher densities of CD3+CD4+ regulatory T cells and all CD3+CD8+ T cell subsets in stromal areas compared to MSI-low/MSS tumors (Figure 2). MSI-high tumors had significantly lower densities of CD3+CD4+ memory T cells in stromal tissue areas compared to MSI-low/MSS.
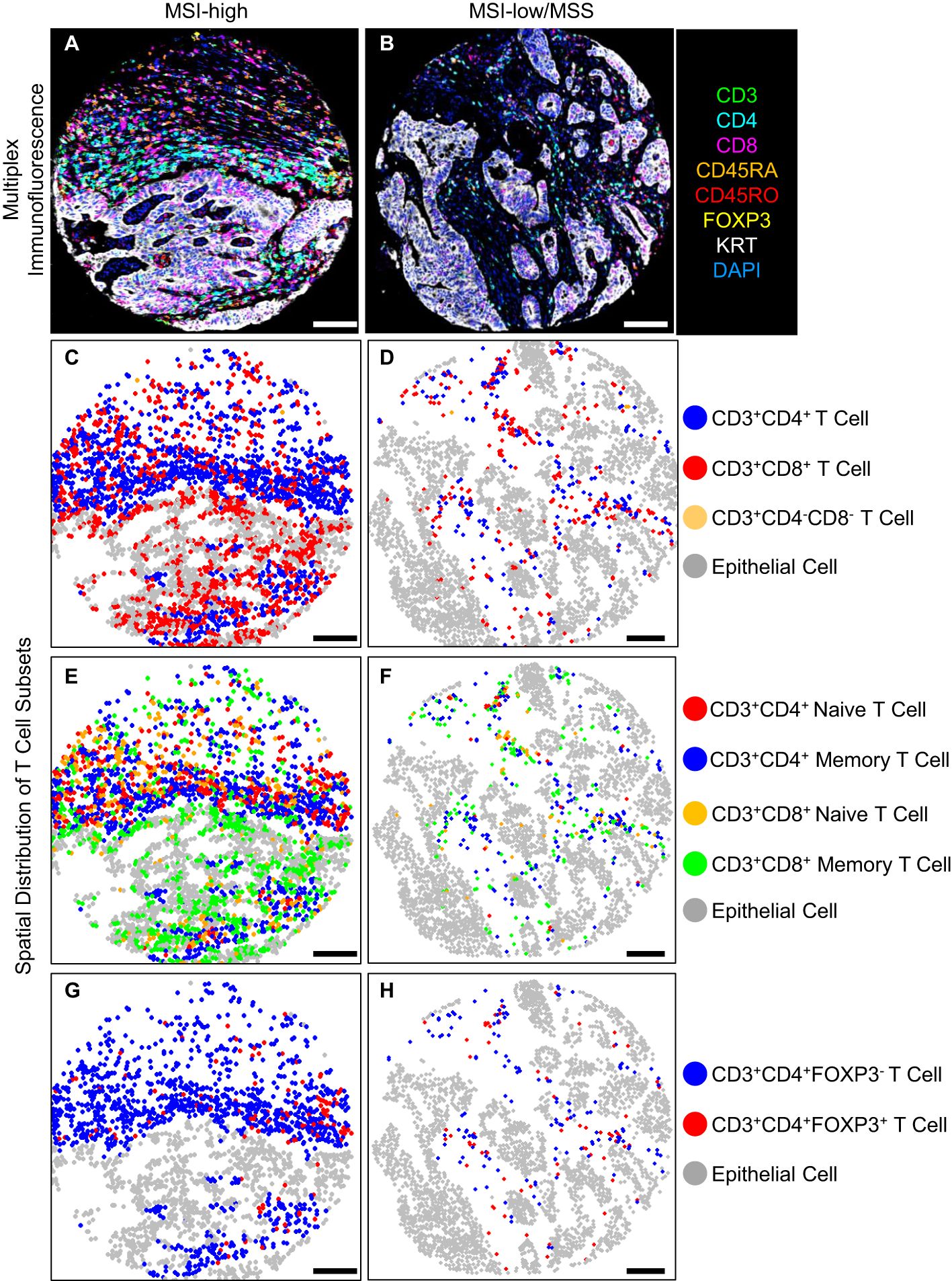
Figure 1. T cell density levels in an MSI-high tumor compared to an MSI-low/MSS tumor, (A, B) multiplex immunofluorescence, (C-H) spatial distribution of T cell subsets, scale bar 100 (μm).
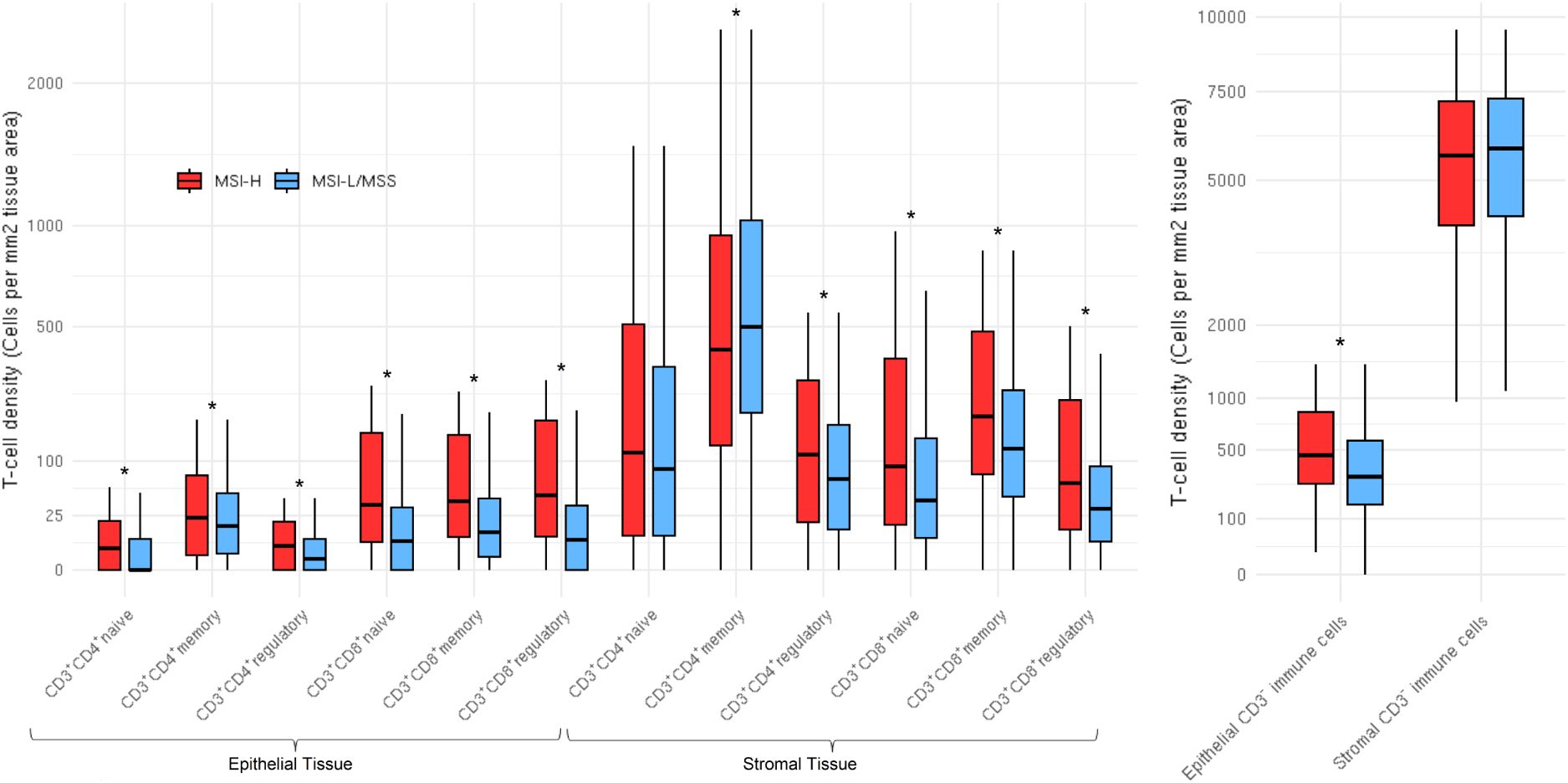
Figure 2. Distributions of T cell subsets and CD3- immune cells stratified by microsatellite instability status in both epithelial (N=1,235) and stromal (N=1,233) tissue areas. CD3+CD4-CD8- (double negatives) distributions not shown due to high percentage of zeros. Distributions are winsorized to their 95th percentile for visualization purposes. The box denotes the interquartile range with a line for median value, and the length of the vertical line represents 1.5 times the smallest value below 25th and 1.5 times the largest value above the 75th percentiles. Stars represent subsets that were significantly different; all subsets were significantly (pFDR < 0.05) different by MSI status, except stromal tissue area CD3+CD4+ naive T cells and stromal tissue area CD3- immune cells.
In ordinal logistic models adjusted for age, sex, study batch, and cancer site, MSI-high status was associated with greater odds of higher density quantile for CD3+CD4+ naive and regulatory subsets, all CD3+CD8+ subsets, double negative T cells, and CD3- non-tumor cells in epithelial tissue areas (Table 2). MSI-high status was also associated with greater odds of higher quantile for CD3+CD4+ regulatory T cells and all CD3+CD8+ subsets in stromal tissue areas, but with lower quantile for stromal area CD3+CD4+ memory T cells [OR per increasing quantile (95% CI) = 0.68 (0.51, 0.91), Table 2]. The strongest associations were for CD3+CD8+ T cells in epithelial areas, where those with MSI-high tumors had around 3 times the odds of greater quantile of epithelial tissue area CD3+CD8+ T cell densities compared to MSI-low/MSS tumors [OR per increasing quantile (95% CI) for CD3+CD8+ naive, memory, and regulatory subsets = 3.49 (2.57, 4.75); 2.82 (2.10, 3.78); 3.04 (2.24, 4.13), respectively].
Associations between MSI status and T cell subsets were generally similar across strata defined by proliferation status, with few exceptions (Table 3). In particular, the association of MSI status with CD3+CD4+ memory T cell densities in epithelial and stromal tissue areas differed by proliferation status. In epithelial tissue areas, MSI-high status was more strongly associated with higher levels of proliferating CD3+CD4+ memory T cells than with non-proliferating CD3+CD4+ memory T cells. In stromal tissue areas, MSI status was more strongly associated with lower levels of non-proliferating CD3+CD4+ memory T cells than proliferating CD3+CD4+ memory T cells. Double negative naive T cells were also differentially associated with MSI status by both tissue area and proliferation status.
T cell density differences by hypermutation statusIn the subset of participants who had hypermutation status available (N=639), 108 individuals were MSI-high, 107 were hypermutated, and 99 were both MSI-high and hypermutated. Among the 639 samples with available data, only 17 were discordant for their MSI-high and hypermutation statuses (Supplementary Table 2). Associations between hypermutation status and T cell subsets were largely consistent with associations found for MSI status (Table 4). Notable exceptions were epithelial area CD3+CD4+ memory and double negative naive T cells: epithelial area CD3+CD4+ memory cells were associated with hypermutation status but not with MSI status [hypermutation status OR (95% CI) = 1.68 (1.11, 2.53), MSI OR = 1.28 (0.96, 1.71)], while epithelial double negative naive T cells were associated with MSI but not hypermutation status [hypermutation status OR (95% CI) = 1.54 (0.46, 4.48), MSI OR = 2.41 (1.16, 4.89), Tables 2 and 4]. When examining total tumor mutational burden (indels and single nucleotide variants) continuously, we observed strong correlations with overall CD3+CD8+ T cells in both epithelial and stromal areas but not with epithelial or stromal area overall CD3+CD4+ T cells (Supplementary Figure 3).
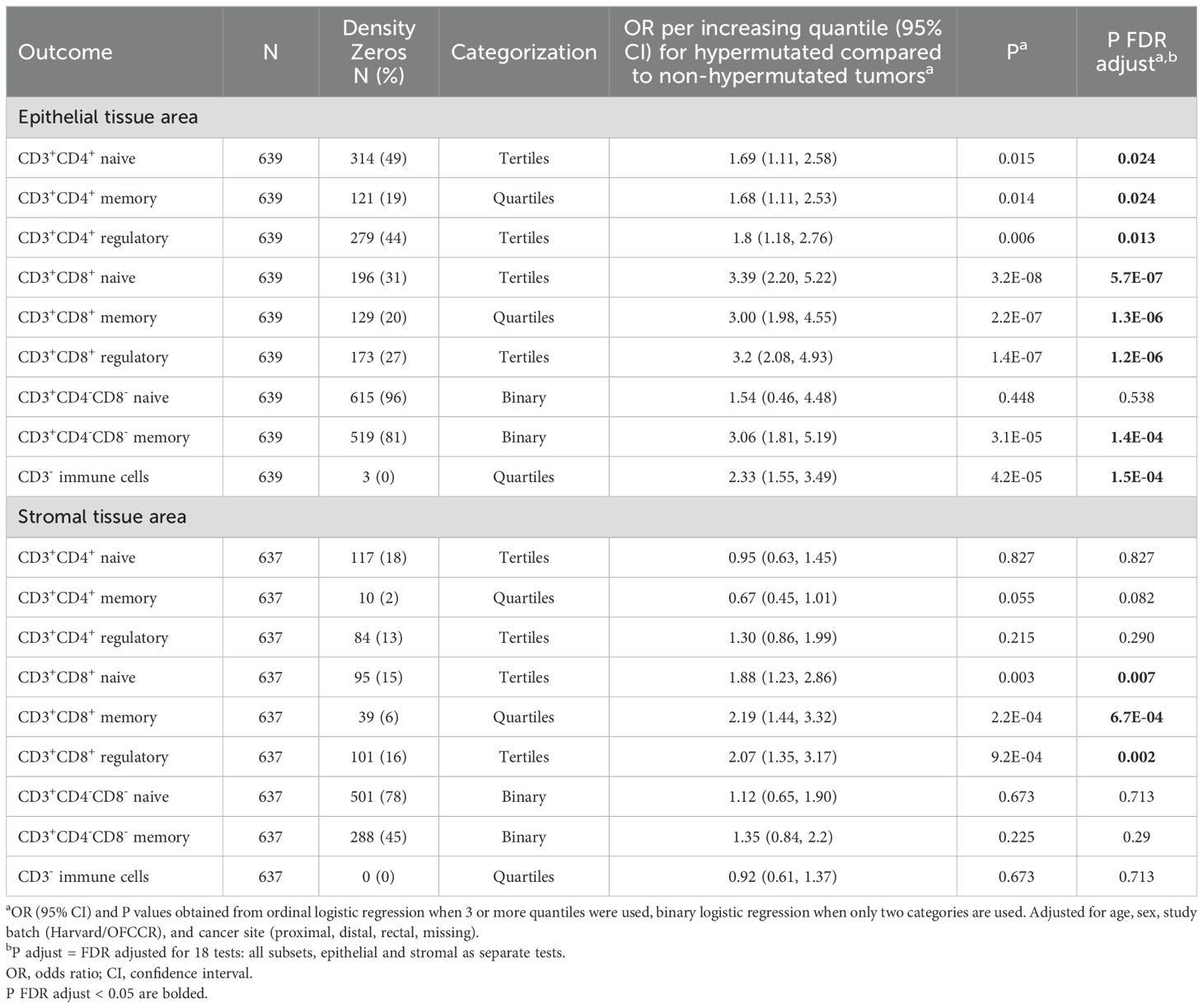
Table 4. Association between hypermutation status and T cell subset densities.
DiscussionIn this large population-based study of T cell densities in CRC, MSI-high status was associated with higher density quantile for the majority of immune subsets in both epithelial and stromal tissue areas. The notable exception was CD3+CD4+ memory T cells in stromal areas, where MSI-high status was significantly associated with lower quantile of that subset compared to MSI-low/MSS tumors. CD3+CD8+ T cells in epithelial areas were particularly strongly associated with MSI-high status. Associations were largely consistent stratifying by proliferation status and examining hypermutation status as the main exposure of interest.
Our results are consistent with previous work, where MSI status is well-known to be associated with overall T cell response in CRC (10–13). Although few studies have examined associations with T cell subsets, at least one prior study noted a specific positive association of CD45RO+ memory T cells with MSI-high status (14). FOXP3+ regulatory T cells have also been found to have higher intraepithelial infiltration in MSI-high tumors compared to in MSS tumors (31). MSI status is a critical variable to examine in CRC, as it reflects a somatic deficiency in DNA mismatch repair resulting in high tumor mutation burden and, therefore, high neoantigen presentation. In particular, MSI-high tumors, with accumulated somatic mutations, may modulate immune response through upregulated expression of immune checkpoints (26). As such, MSI status is also known to be strongly associated with treatment response, particularly treatment with immune checkpoint inhibitors (4–6), and survival (32–34). Interestingly, a recent meta-analysis of 13,029 patients combining both MSI status and tumor infiltrating lymphocytes (TILs) found that better survival was only observed among CRC patients that were TILs-high, regardless of MSI status- although patients with MSI-high TILs-low did still have better survival than those MSS TILs-low, particularly for CRC-specific survival (35). This study supports efforts to include both T cell densities and MSI status in clinical decision making.
Our study examines the association between MSI status and T cell specific subset densities within both epithelial and stromal tissue. We prioritized identifying non-overlapping subsets of naive, memory, and regulatory both CD3+CD4+ and CD3+CD8+ subsets of T cells due to the different roles these cell types play in the tumor immune microenvironment. For example, CD3+CD4+ T cells support the expansion and differentiation of CD3+CD8+ T cells, as well as conducting surveillance, regulating tissue homeostasis, and restricting adaptive immune response (36). In contrast, CD3+CD8+ T cells kill neoplastic cells and pathogens to drive anti-cancer immune response (37). T cells are functionally considered naive until they encounter their specific antigen in peripheral tissues, after which they differentiate into either effector or memory T cells (38). Naïve and memory T cells have been found to have different chromatin accessibility and transcription factor expression (38). Regulatory T cells, primarily CD3+CD4+FOXP3+ T cells, are involved in immune homeostasis and auto-immunity prevention (39). In addition to differential roles of different subsets, the location of T cells is also critical and influences function (40). For example, only T cells within tumor epithelial areas, versus stromal areas, can eliminate tumor cells through direct cell-cell contact (40–43). The heterogeneity of the tumor immune microenvironment, both through cell function and location, indicate the importance of examining associations critically between immune cell populations and predictor or outcomes of interest. For example, in our analysis we found that MSI-high tumors had 2 times the odds of greater quantile of epithelial tissue area CD3+CD4+ regulatory T cell densities compared to MSI-low/MSS tumors, which may have been obscured if CD3+CD4+ T cell densities were examined overall. Examining immune subsets in broad, generalized categories may obscure subset-specific associations that are informative of immune response biology.
Although the majority of T cell subsets were observed to be positively associated with MSI-high status in our analysis, we also found that MSI-high tumors had lower quantile densities of CD3+CD4+ memory T cells in stromal areas. Memory CD3+CD4+ T cells are derived from a cell population that experienced a specific antigen but remained in the body after said antigen was no longer present, where they may be protective against cancer but also be involved in auto-immunity, allergy, and chronic inflammation (36). Although the basis for our observed inverse finding between density of this subset and MSI-high status is not clear, this unexpected association may be influenced by other immune cells in the tumor microenvironment that were not profiled as a part of our study- for example, macrophages or dendritic cells. Further research is needed on how MSI-high status impacts memory CD3+CD4+ T cells in stromal tissue regions.
The differential responsiveness of MSI-high compared to MSI-low/MSS CRCs to immune checkpoint blockade treatment provides strong evidence that MSI status is a clinically relevant tumor characteristic (44). While MSI-high tumors clearly had statistically significantly higher T cell densities for many subsets in our study, it is important to note that the absolute difference between MSI-high and MSI-low/MSS is relatively modest. MSI-low/MSS tumors still had reasonable densities of many T cell populations that were not orders of magnitude different from those observed in MSI-high tumors. Moreover, the previously mentioned meta-analysis combining both TILs and MSI status in relation to survival found that the MSS TILs-high subtype had CRC-specific survival hazard ratios that were very comparable to those of MSI-high TILs-high (HR=0.55, 0.47-0.64 and HR=0.53, 0.43-0.66, respectively) (35), suggesting that MSS tumors with high levels of T cell densities may behave more similarly to MSI-high tumors. This finding may provide motivation for future treatments to better exploit the T cell response present in MSI-low/MSS CRC tumors.
Our study has notable strengths. This is the first study, to our knowledge, to examine the association of MSI and hypermutation status with specific non-overlapping T cell subsets using a mIF assay, where we have the ability to determine non-overlapping specific naive, memory, and regulatory subsets of cytotoxic and helper T cells in both epithelial and stromal tissue areas, as well as double negative T cells and CD3- immune cells. Our integrated CRC tumor databases (15, 16) enabled us to link MSI and hypermutation status with the features of the tumor microenvironment while accounting for demographic and epidemiologic variables. We also acknowledge several limitations in this study. The vast majority of our sample was non-Hispanic White individuals, necessitating future studies in other populations. Additionally, while our study is notable in its inclusion of specific T cell density subsets that include differentiation by naive, memory, or regulatory subtype, as well as proliferation status- our study only includes information on T cell densities (i.e., cell count per mm2) and broad tissue location (i.e., tumor intraepithelial vs. stromal regions), rather than additionally including information on spatial biology of the tumor. Spatial organization of the tumor immune microenvironment has been shown to be independently prognostic of CRC outcomes (45–49). In future work, detailed data on the spatial biology from both TMAs as well as whole slides will allow us to more comprehensively address this question of spatial organization of the tumor immune microenvironment in relation to survival and tumor characteristics. Previous work has suggested that assessment of tumor structures is more powerfully addressed through whole-slide imaging, rather than TMAs (50). We acknowledge the limitation of our study in using TMAs to examine the tumor microenvironment, however, TMA sections contain relevant information and vastly improve researchers ability to conduct studies of immune profiling in large scale population studies, where multiplex imaging analysis of whole slide images is cost prohibitive. Additionally, the use of TMAs expanding our sample size allows us to examine different subset breakdowns, such as quartiles of T cell densities among MSI-high tumors, which we may not be able to examine otherwise with smaller numbers due to low statistical power. Furthermore, in future work we aim to compare whole slide imaging with our TMA results to compare our findings utilizing different methods. In future work we also aim to examine mechanisms of immune cell recruitment and immune checkpoint molecules as these are important pathways that are likely at play in the relationship between MSI status and T cell densities.
In conclusion, MSI-high and hypermutation status were strongly associated with different T cell and immune subsets in both epithelial and stromal tissue areas, where the strongest associations were present for CD3+CD8+ T cells in epithelial tissue areas. Our large-scale investigation indicates potential mechanisms by which MSI-high status confers better survival and supports ongoing and future efforts to identify patients who may be more likely to respond to immunotherapy treatments.
Data availability statementThe raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statementAll participants gave written informed consent and studies were approved by their respective Institutional Review Boards. The Nurses’ Health Study and Health Professionals Follow-up Study protocols were approved by the institutional review boards of the Brigham and Women’s Hospital and Harvard T.H. Chan School of Public Health, and those of participating registries as required. All studies were conducted in accordance with the local legislation and institutional requirements.
Author contributionsCT: Conceptualization, Formal analysis, Investigation, Methodology, Software, Supervision, Visualization, Writing – original draft, Writing – review & editing. YT: Conceptualization, Data curation, Investigation, Resources, Software, Validation, Writing – review & editing. EW: Conceptualization, Formal analysis, Methodology, Software, Writing – review & editing. TU: Conceptualization, Data curation, Funding acquisition, Investigation, Resources, Writing – review & editing. RS: Data curation, Project administration, Resources, Writing – review & editing. DB: Conceptualization, Investigation, Writing – review & editing. CQ: Data curation, Project administration, Resources, Writing – review & editing. LH: Formal analysis, Writing – review & editing. AC: Data curation, Investigation, Writing – review & editing. SG: Resources, Writing – review & editing. RG: Resources, Writing – review & editing. JH: Conceptualization, Writing – review & editing. ST: Data curation, Investigation, Project administration, Writing – review & editing. SO: Conceptualization, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Writing – review & editing. AP: Conceptualization, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Writing – review & editing. JN: Conceptualization, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Writing – review & editing. UP: Conceptualization, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Writing – review & editing.
FundingThe author(s) declare financial support was received for the research, authorship, and/or publication of this article. Genetics and Epidemiology of Colorectal Cancer Consortium (GECCO): National Cancer Institute, National Institutes of Health, U.S. Department of Health and Human Services (U01 CA137088, R01 CA059045, U01 CA164930, R21 CA191312, R01 CA244588, R01 CA206279, R01 CA201407, R01 CA248857, P20 CA252733). Genotyping/Sequencing services were provided by the Center for Inherited Disease Research (CIDR) contract number HHSN268201700006I and HHSN268201200008I. This research was funded in part through the NIH/NCI Cancer Center Support Grant P30 CA015704. Scientific Computing Infrastructure at Fred Hutch funded by ORIP grant S10OD028685. CT is supported by L70 CA284301, T32 CA094880, and T32 CA009168. DB is supported by a National Health and Medical Research Council of Australia Investigator grant (GNT1194896) and the University of Melbourne Dame Kate Campbell Fellowship. The Colon Cancer Family Registry (CCFR, www.coloncfr.org) is supported in part by funding from the National Cancer Institute (NCI), National Institutes of Health (NIH) (award U01 CA167551). Support for case ascertainment was provided in part from the Surveillance, Epidemiology, and End Results (SEER) Program and the following U.S. state cancer registries: AZ, CO, MN, NC, NH; and by the Victoria Cancer Registry (Australia) and Ontario Cancer Registry (Canada). Additional support for OFCCR/ARCTIC was through NCI/NIH awards U01/U24 CA074783 (to SG). Additional funding for the genetic analyses of OFCCR/ARCTIC was through award GL201-043 from the Ontario Research Fund (to BWZ), award 112746 from the Canadian Institutes of Health Research (to TJH), through a Cancer Risk Evaluation (CaRE) Program grant from the Canadian Cancer Society, and through generous support from the Ontario Ministry of Research and Innovation. Nurses’ Health Study, Health Professionals Follow-up Study: HPFS was supported by the National Institutes of Health (P01 CA055075, UM1 CA167552, U01 CA167552, R01 CA151993, R35 CA197735), NHS by the National Institutes of Health (P01 CA087969, UM1 CA186107, R01 CA151993, R35 CA197735). TU was supported by grants from the USA National Institutes of Health (R50 CA247122), the Prevent Cancer Foundation, Brigham and Women’s Hospital, Harvard T.H. Chan School of Public Health, and American Institute for Cancer Research. SO was additionally supported by USA National Institutes of Health grants (R35 CA197735, R01 CA248857), the Cancer Research UK Grand Challenge Award (C10674/A27140) and the American Cancer Society Clinical Research Professor Award (CRP-24-1185864-01-PROF).
AcknowledgmentsThe Colon Cancer Family Registry (CCFR): The Colon CFR graciously thanks the generous contributions of their study participants, dedication of study staff, and the financial support from the U.S. National Cancer Institute, without which this important registry would not exist. We acknowledge Channing Division of Network Medicine, Department of Medicine, Brigham and Women’s Hospital as home of the NHS. The authors would like to acknowledge the contribution to this study from central cancer registries supported through the Centers for Disease Control and Prevention’s National Program of Cancer Registries (NPCR) and/or the National Cancer Institute’s Surveillance, Epidemiology, and End Results (SEER) Program. Central registries may also be supported by state agencies, universities, and cancer centers. Participating central cancer registries include the following: Alabama, Alaska, Arizona, Arkansas, California, Colorado, Connecticut, Delaware, Florida, Georgia, Hawaii, Idaho, Indiana, Iowa, Kentucky, Louisiana, Massachusetts, Maine, Maryland, Michigan, Mississippi, Montana, Nebraska, Nevada, New Hampshire, New Jersey, New Mexico, New York, North Carolina, North Dakota, Ohio, Oklahoma, Oregon, Pennsylvania, Puerto Rico, Rhode Island, Seattle SEER Registry, South Carolina, Tennessee, Texas, Utah, Virginia, West Virginia, Wyoming. The authors assume full responsibility for analyses and interpretation of these data.
Conflict of interestRG received a graduate scholarship from Pfizer and provided consulting or advisory roles for Astrazeneca, Tempus, Eisai, Incyte, Knight Therapeutics, Guardant Health, and Ipsen, unrelated to this manuscript. JN received consulting fees from Leica Biosystems and research grant support from Natera. UP was a consultant with AbbVie and her husband is holding individual stocks for the following companies: BioNTech SE – ADR, Amazon, CureVac BV, NanoString Technologies, Google/Alphabet Inc Class C, NVIDIA Corp, Microsoft Corp.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statementThe author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Author disclaimerThe content of this manuscript does not necessarily reflect the views or policies of the NCI, NIH or any of the collaborating centers in the Colon Cancer Family Registry (CCFR), nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government, any cancer registry, or the CCFR.
Supplementary materialThe Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2024.1505896/full#supplementary-material
Supplementary Table 1 | Antibodies and Staining Conditions for Multiplex Immunofluorescence Histological Analysis.
Supplementary Table 2 | Participant characteristics overall and by MSI status. *MSI = microsatellite instability high (MSI-high), low (MSI-low), or microsatellite stable (MSS). P values were calculated using t test for mean differences & chi square for frequencies.
Supplementary Figure 1 | (A) Profiling of the multiplexed immunofluorescence panel, utilizing antibodies targeting CD3, CD4, CD8, CD45RA, CD45RO, FOXP3, KRT, and DAPI. (B-E). Multiplex immunofluorescence images (B, C) were analyzed to classify both tissue and cellular categories. Tissue segmentation (D) was performed to delineate epithelial (red), stromal (green), and other (blue) regions. Cell segmentation (E) was then sequentially applied to classify individual cells. Scale bar: 100 (μm). (F) Identification of distinct T-cell subsets via the co-expression of T-cell markers (membrane CD3, membrane CD4, membrane CD8, membrane CD45RA, membrane CD45RO, nucleus FOXP3), epithelial marker (cytoplasm KRT), and DNA marker (nucleus DAPI) at single-cell resolution.
Supplementary Figure 2 | Boxplot and beeswarm plot distributions of (A) T cell subsets and (B) CD3- immune cells stratified by epithelial (N=1,235) and stromal (N=1,233) tissue area. CD3+CD4-CD8- (double negatives) distributions not shown due to high percentage of zeros. The box denotes the inter-quartile range with a line for median value, and the length of the vertical line represents 1.5 times the smallest value below 25th and 1.5 times the largest value above the 75th percentiles.
Supplementary Figure 3 | Scatter plots and correlation of tumor mutational burden and T cell densities. Footnote: N = 639 in epithelial areas, N = 637 in stromal areas.
References1. Guinney J, Dienstmann R, Wang X, de Reyniès A, Schlicker A, Soneson C, et al. The consensus molecular subtypes of colorectal cancer. Nat Med. (2015) 21:1350–6. doi: 10.1038/nm.3967
PubMed Abstract | Crossref Full Text | Google Scholar
2. Zheng Z, Wieder T, Mauerer B, Schäfer L, Kesselring R, Braumüller H. T cells in colorectal cancer: unravelling the function of different T cell subsets in the tumor microenvironment. Int J Mol Sci. (2023) 24:11673. doi: 10.3390/ijms241411673
PubMed Abstract | Crossref Full Text | Google Scholar
3. Taieb J, Svrcek M, Cohen R, Basile D, Tougeron D, Phelip JM. Deficient mismatch repair/microsatellite unstable colorectal cancer: Diagnosis, prognosis and treatment. Eur J Cancer Oxf Engl 1990. (2022) 175:136–57. doi: 10.1016/j.ejca.2022.07.020
PubMed Abstract | Crossref Full Text | Google Scholar
4. Tan E, Sahin IH. Defining the current role of immune checkpoint inhibitors in the treatment of mismatch repair-deficient/microsatellite stability-high colorectal cancer and shedding light on future approaches. Expert Rev Gastroenterol Hepatol. (2021) 15:735–42. doi: 10.1080/17474124.2021.1886077
PubMed Abstract | Crossref Full Text | Google Scholar
5. Picard E, Verschoor CP, Ma GW, Pawelec G. Relationships between immune landscapes, genetic subtypes and responses to immunotherapy in colorectal cancer. Front Immunol. (2020) 11:369. doi: 10.3389/fimmu.2020.00369
PubMed Abstract | Crossref Full Text | Google Scholar
6. André T, Shiu K-K, Kim TW, Jensen BV, Jensen LH, Punt C, et al. Pembrolizumab in microsatellite-instability–high advanced colorectal cancer. N Engl J Med. (2020) 383:2207–18. doi: 10.1056/NEJMoa2017699
PubMed Abstract | Crossref Full Text | Google Scholar
7. Yuza K, Nagahashi M, Watanabe S, Takabe K, Wakai T. Hypermutation and microsatellite instability in gastrointestinal cancers. Oncotarget. (2017) 8:112103–15. doi: 10.18632/oncotarget.22783
PubMed Abstract | Crossref Full Text | Google Scholar
10. Alexander J, Watanabe T, Wu TT, Rashid A, Li S, Hamilton SR. Histopathological identification of colon cancer with microsatellite instability. Am J Pathol. (2001) 158:527–35. doi: 10.1016/S0002-9440(10)63994-6
PubMed Abstract | Crossref Full Text | Google Scholar
11. Shia J, Ellis NA, Paty PB, Nash GM, Qin J, Offit K, et al. Value of histopathology in predicting microsatellite instability in hereditary nonpolyposis colorectal cancer and sporadic colorectal cancer. Am J Surg Pathol. (2003) 27:1407–17. doi: 10.1097/00000478-200311000-00002
PubMed Abstract | Crossref Full Text | Google Scholar
12. Jenkins MA, Hayashi S, O’Shea AM, Burgart LJ, Smyrk TC, Shimizu D, et al. Pathology features in Bethesda guidelines predict colorectal cancer microsatellite instability: a population-based study. Gastroenterology. (2007) 133:48–56. doi: 10.1053/j.gastro.2007.04.044
PubMed Abstract | Crossref Full Text | Google Scholar
13. Ogino S, Nosho K, Irahara N, Meyerhardt JA, Baba Y, Shima K, et al. Lymphocytic reaction to colorectal cancer is associated with longer survival, independent of lymph node count, microsatellite instability, and CpG island methylator phenotype. Clin Cancer Res Off J Am Assoc Cancer Res. (2009) 15:6412–20. doi: 10.1158/1078-0432.CCR-09-1438
PubMed Abstract | Crossref Full Text | Google Scholar
14. Nosho K, Baba Y, Tanaka N, Shima K, Hayashi M, Meyerhardt JA, et al. Tumour-infiltrating T-cell subsets, molecular changes in colorectal cancer, and prognosis: cohort study and literature review. J Pathol. (2010) 222:350–66. doi: 10.1002/path.v222:4
Crossref Full Text | Google Scholar
15. Newcomb PA, Baron J, Cotterchio M, Gallinger S, Grove J, Haile R, et al. Colon Cancer Family Registry: an international resource for studies of the genetic epidemiology of colon cancer. Cancer Epidemiol biomark Prev Publ Am Assoc Cancer Res Cosponsored Am Soc Prev Oncol. (2007) 16:2331–43. doi: 10.1158/1055-9965.EPI-07-0648
PubMed Abstract | Crossref Full Text | Google Scholar
16. Zhao M, Lau MC, Haruki K, Väyrynen JP, Gurjao C, Väyrynen SA, et al. Bayesian risk prediction model for colorectal cancer mortality through integration of clinicopathologic and genomic data. NPJ Precis Oncol. (2023) 7:57. doi: 10.1038/s41698-023-00406-8
留言 (0)