
記住我
Interstitial lung diseases (ILDs) are a heterogeneous group of lung diseases characterised by fibrosis (scarring) of the parenchyma. ILD classification is based on histopathological, radiologic, aetiologic, and clinical criteria (1–3). Idiopathic pulmonary fibrosis (IPF) is a rare, chronic, and progressive fibrosing interstitial pneumonia occurring mainly in the elderly population, with greater incidence after the age of 60 and characterised by a highly poor prognosis (4). Although the aetiology of this disease remains largely unexplored, IPF is the most common of ILD, and its incidence appears to be increasing (5, 6). IPF is characterised by irreversible loss of lung function due to fibrosis, manifesting as chronic cough and progressive worsening of dyspnoea, with a median survival from diagnosis of approximately 2–4 years. However, it is difficult to predict the clinical course due to the significant variability among cases. Lung cancer (LC) is one of the most significant comorbidities occurring in patients with ILDs, mainly IPF (7). It is well established that the two diseases share common risk factors (smoking habits) and some molecular traits (8–10). The combination of the two pathologies is a severe condition, with a negative impact on both mortality and the quality of life of patients (11, 12). It is therefore essential to structure a therapeutic strategy that improves survival prospects, as well as the quality of life of these patients (13). Unfortunately, no standardised diagnostic and therapeutic approaches have been defined for these neglected cases. Sometimes, a palliative approach is the only option. It is, thus, mandatory to identify parameters/variables that can help in patient stratification and outcome prediction to identify effective strategies for managing concomitant diseases. The rationale of the present work is to analyse demographic and clinical data from a cohort of IPF patients with concomitant lung cancer, aiming to rank the various risk factors and identify mortality predictive markers.
2 Patients and methods2.1 Patient identification and selectionThe study retrospectively analyses data from a consecutive series of patients who were observed at three reference ILD and thoracic oncology centres in Italy: the IRCCS San Matteo Hospital Foundation in Pavia, the Azienda Aspedaliera Specialistica dei Colli in Naples, and the Policlinico Riuniti in Foggia. Data collection spans from the beginning of 2018 to the end of 2023. This is not a clinical trial. Exhaustive clinical reports regarding the evaluated population are available in Table 1. Informed consent from each patient was routinely collected at hospital admission in accordance with standard hospital procedures. Patient data were collected through consultation of operating directories; oncological, pneumological, and pathological reports; and discharge letters.
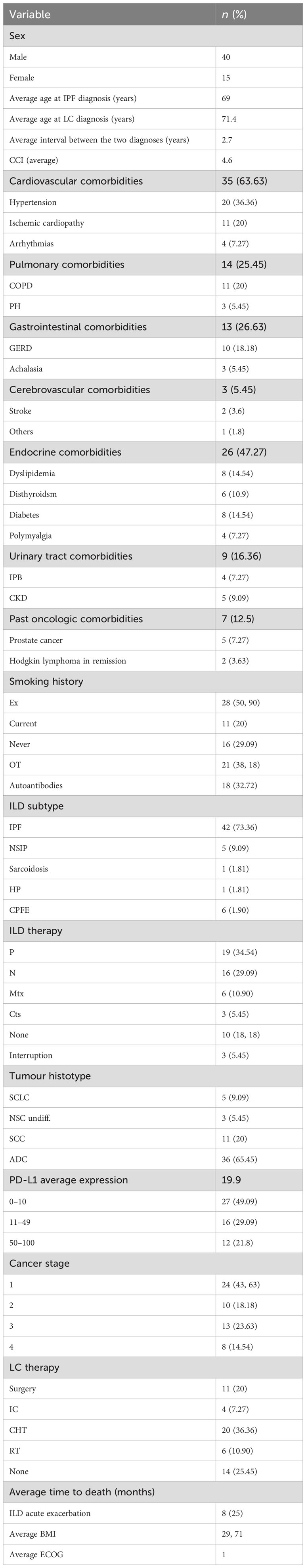
Table 1. Demographic and clinical features of the evaluated population.
2.2 Immunohistochemistry and geneticsTumour samples were obtained from patients who underwent surgical resection or biopsy. The sample was considered eligible for the study if the tumour morphology was preserved and a minimum of 100 cancer cells were present in the tissue section. Morphologic classification was assigned according to the last World Health Organisation lung cancer classification criteria (14), and the primary site of origin was confirmed by the immunohistochemical phenotype (thyroid transcription factor-1, p40 (15, 16)) by the two senior lung pathologists. According to recent guidelines (17), formalin-fixed paraffin-embedded (FFPE) tumour samples were retrospectively selected and submitted to immunohistochemical assay or analysis of PD-L1 (programmed death ligand 1) expression levels using a laboratory-developed test designed to optimise the use of the anti-PD-L1 22C3 antibody (Dako) on the Omnis autostainer (Dako, AGILENT technologies, Santa Clara (CA), USA) with the Envision FLEX (Dako) revelation system. PD-L1 IHC using the PD-L1 IHC 22C3 pharmDx kit was performed according to manufacturer recommendations (website at http://www.ventana.com/ventana-pd-l1-sp263-assay-2/). The IHC protocol was performed on specimens sectioned at a thickness of 3 μm and stained on positively charged glass slides stored at 4°C within 3 days after sectioning, according to already published protocols (18). Tumour proportion score (TPS) was evaluated according to published guidelines (14). PD-L1 expression was classified into TPS < 1% (negative), TPS 2% to 5% (weak), TPS 6% to 49% (positive), and TPS ≥ 50% (strong) (19–21). To assess the tumour mutational profile, tumour DNA was extracted using the DNA FFPE tissue kit (Omega, Norcross, GA, USA) according to the guidebook, and concentrations were detected by Qubit® 2.0 fluorometer dsDNA HS assay kit (Thermo Fisher Scientific Inc., Waltham, MA, USA). Genetic analysis was performed according to routine procedures and widely described protocols (22–24), based on Next-Generation Sequencing (NGS) technology targeting 94 genes and 284 single-nucleotide polymorphisms.
2.3 Statistical analysisStatistical analysis was carried out using the Excel add-in package. The continuous variables were expressed as the mean value ± the standard deviation (SD), and the latter was compared using the Student’s t-test for independent variables. The nominal variables were compared using χ2 tests. The Kaplan–Meier method was used to generate the cumulative survival curves, and the log-rank test was used to calculate the differences between the curves. A p-value < 0.05 was considered statistically significant. The entire dataset was then tested using the JMP partition algorithm (JMP-Statistical Discoveries, from SAS; website at www.jmp.com), which can search for all possible subdivisions of the best predictors of response/outcome and event probability distribution.
3 Results3.1 Demographic and clinical annotationLung cancer associated with ILDs is confirmed to be a rare condition, representing 2.1% of the total LC diagnoses in the three clinical centres during the study interval, whereas it aroused with an average interval of 2.7 years after the ILD diagnosis. Overall, the study screened and identified 73 patients; of them, 17 cases were excluded from the analysis due to the lack of complete clinicopathologic data, and one case was diagnosed with pleural mesothelioma. The remaining 55 ILD+LC patients were evaluated; their exhaustive features are reported in Table 1, while the study design is described in Figure 1. In the analysed population, most diagnoses (about 74% of cases) referred to IPF, while the remaining cases were diagnosis with nonspecific interstitial pneumonia (NSIP) and combined pulmonary fibrosis and emphysema (CFPE) in about 10% of cases. LC was associated with sarcoidosis in one case and hypersensitivity pneumonitis (HP) in another. The diagnosis of various ILDs was performed through the integration of anamnestic, clinicopathologic, radiological, and serological data, along with a multidisciplinary discussion, according to evidence-based diagnostic guidelines (25–27). An extended panel of autoantibody testing was applied to the blood of each case evaluated (28, 29). Autoantibodies were tested by immunoprecipitation, and all were IgG, except for the rheumatoid factor, which belongs to the IgM class by definition. Most ILD patients were treated with antifibrotic agents: 19 patients (35%) were treated with pirfenidone, 16 (29%) with nintedanib, six (11%) with methotrexate, and three (3.7%) with oral steroids. Male patients and smokers are at higher risk of developing both diseases (72.72% and 70.9% of cases, respectively). The pathogenic role of smoking is also coherent with the high incidence (63.63%) of cardiovascular illness in the analysed cohort. Similarly, 20% of patients had functional parameters of airway obstruction and/or static hyperinflation consistent with COPD. Respiratory function tests were performed periodically, with a focus on the analysis conducted at cancer diagnosis. Severe impairment of carbon monoxide diffusion was reported, with an average value of 50.6% of the expected (standard deviation [SD]: 17.8). For lung cancer detection, most cases (47, 85.45%) were diagnosed in stages I to IIIa LC, possibly reflecting the strict computed tomography follow-up for the ILD. A proliferative pattern of transformed cells was selectively limited to the fibrotic context. Indeed, in our cohort, 87% of cancer nodules were detected in the peripheral parenchyma in dense fibrotic areas. LC diagnosis was reached, in the vast majority of cases, through transthoracic CT-guided biopsy, and each case was evaluated by both interdisciplinary boards for thoracic neoplasms and for interstitial and rare lung diseases. Quite independently of the LC stage, patients’ overall survival after cancer diagnosis was 18.8 months, and 17.4 months in the IPF-only population (p-value 0.04). The expression of PD-L1 in tumour cells was extremely heterogeneous and overall positive, with an average of about 20%. With respect to cancer treatment decisions, they were based on interdisciplinary discussion, and it is not surprising that, due to the difficulty in managing these rare patients, treatment in some instances was not fully adherent to current lung cancer guidelines. Radiotherapy has generally been excluded to avoid further damage to the lung’s fibrotic parenchyma surrounding the tumour mass. Consequently, systemic cytotoxic chemotherapy was considered the best treatment choice, even in the early stages. On the other hand, the 25% of cases who received any treatment largely exceeds the 15% of those carrying advanced cancer diagnosis; this is mainly related to the worse performance status associated with concomitant respiratory failure. Overall, 10 patients were excluded from active anticancer therapy due to comorbidities and/or lung function severe impairment; three cases discontinued the initial treatment due to worsening performance status.
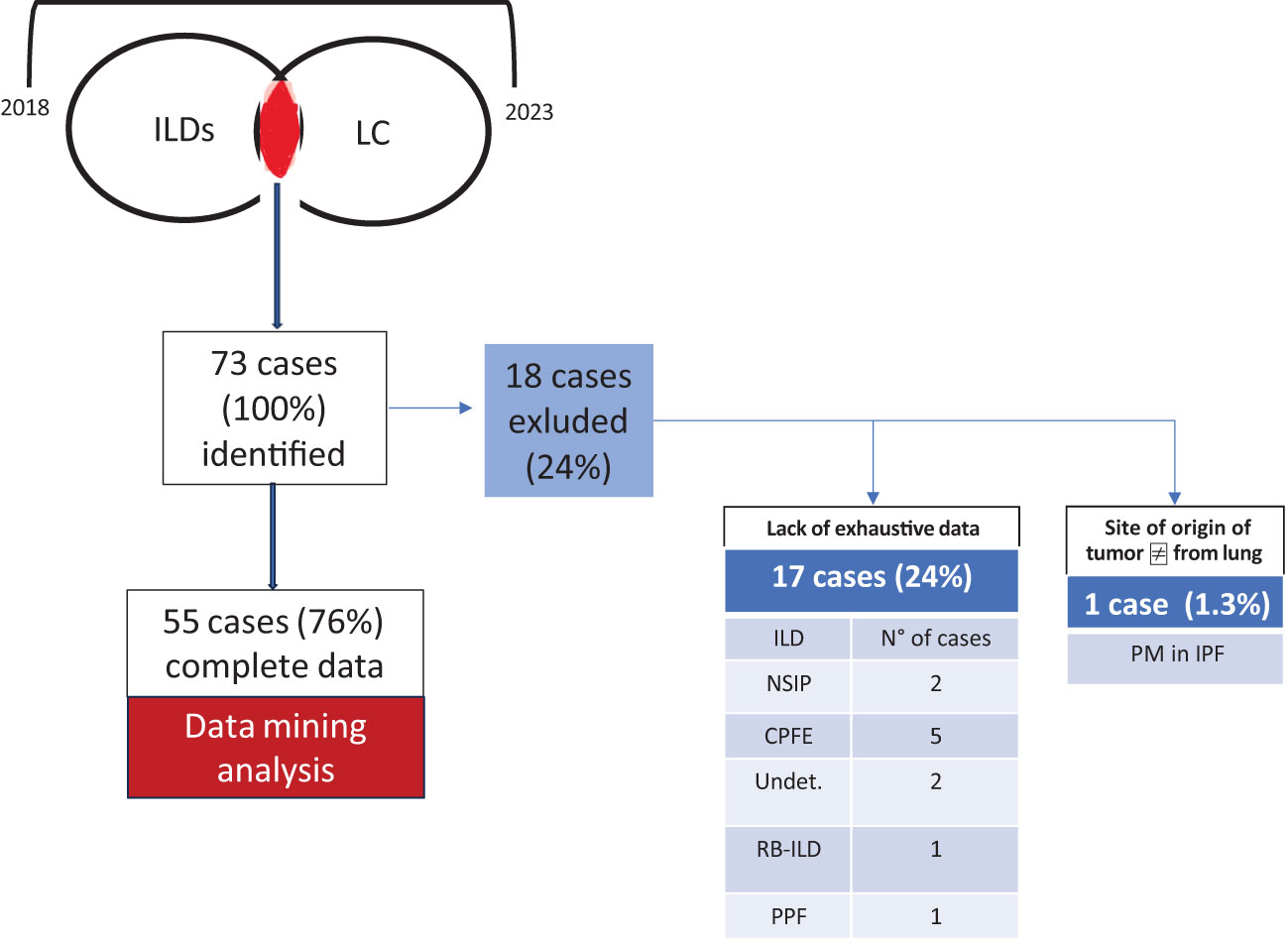
Figure 1. Study design. Overall, 73 cases were identified during the study interval (2018–2023); 18 of them were excluded due to a lack of exhaustive data and the absence of confirmed lung origin of the tumour. CTD, connective tissue disease-associated ILD; undet, undetermined fibrotic disease; RB-ILD, respiratory bronchiolitis interstitial lung disease; PM, pleural mesothelioma. One patient satisfied criteria for progressive pulmonary fibrosis (PPF) according to current guidelines. Serum autoantibodies detected in CTD patients included antinuclear antibodies (ANA) (all patients, 100%) and antibodies against topoisomerase 1 (Scl-70) (two patients, 33%). The patient with “undeterminate UIP” was treated with nintedanib due to a progressive fibrosing course. Patients with CTD-ILD received appropriate immunosuppressive treatment: methotrexate (50% of cases), mycophenolate (9%), and azathioprine (18%).
3.2 Outcome predictorsWe then proceeded to analyse the entire dataset with the aim of identifying the best predictors of patients’ outcomes. To obtain more reliable findings, the homogeneity of the dataset was increased by performing statistical analysis on the IPF cohort alone, the widest ILD population (42 out of 55 cases, 76.4%). Based on this, the row overall survival curve showed differences based on patients’ gender, with women having a lower survival rate than men (p-value < 0.05) (Figure 2A). Within the limits of the cohort in the study, the characteristics of female patients were similar to those of male patients, with the exception of a lower age at IPF diagnosis (61.25 years vs. 70.47 in the IPF cohort), and treatment options, as none of the women underwent surgery, regardless of clinical performance variables and cancer stage. On this evidence, we analysed IPF patients’ outcomes based on the therapy they received. The distribution of survival probabilities was analysed across the five groups representing the different first-line therapeutic strategies. This analysis highlights that the highest probability of survival occurs in the sub-population that underwent surgery for cancer (Figures 2B, C). With respect to the clinical variables that could impact patients’ survival, two conditions emerged as the most relevant. The first factor regards the co-existence of previous diagnoses of COPD, which, in the analysed cohort, acts as a protective factor and is associated with a higher survival probability (Figure 3A). Indeed, the overall survival of patients with associated obstructive conditions is 26 months. All except one of these patients were referred for chronic respiratory failure with the need for oxygen supplementation, and none of them underwent surgery. This point is of extreme interest since, although these patients were not referred for surgery, they were mainly treated with radiotherapy and chemotherapy. Notably, these tumours exhibited weak expression of PD-L1 (TPS% 22), suggesting that the immune reaction associated with COPD may have a protective effect. The lower number of CPFE cases does not allow for a proper comparison. However, within this limitation, patients with CPFE presented lower survival rates (13.3 months) in the absence of significant PD-L1 expression (TPS%: 20.7). The other clinical parameter independently associated with reduced survival is the presence of serum circulating autoantibodies (Figure 3B). None of the enrolled patients were referred for autoimmune disease diagnosis, nor was it confirmed after multidisciplinary revision.
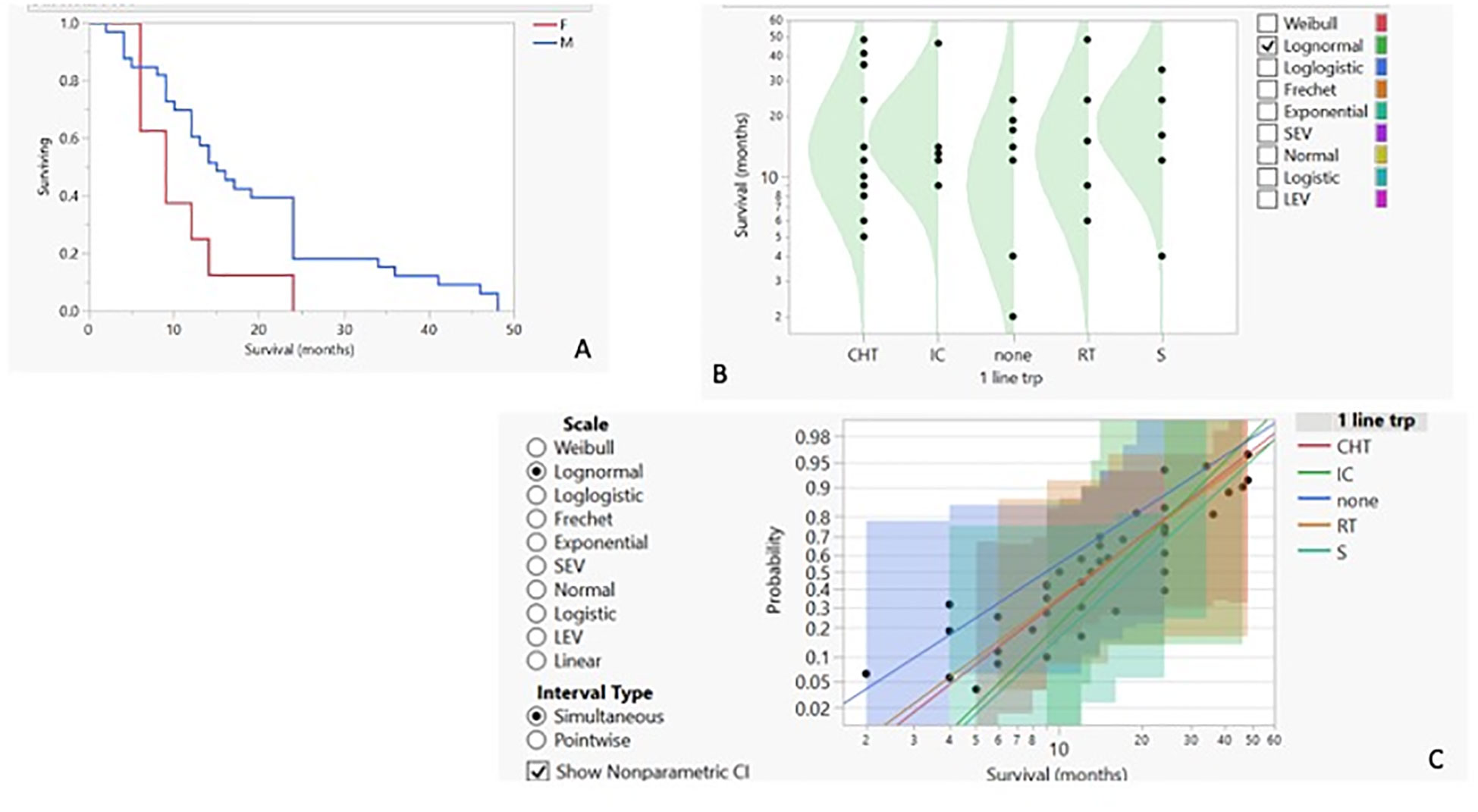
Figure 2. (A) Kaplan–Meier survival curves for IPF patients with LC based on their gender. Survival by first-line therapy against cancer: probability density curve (B) with logarithmic fit–nonparametric overlay (C).
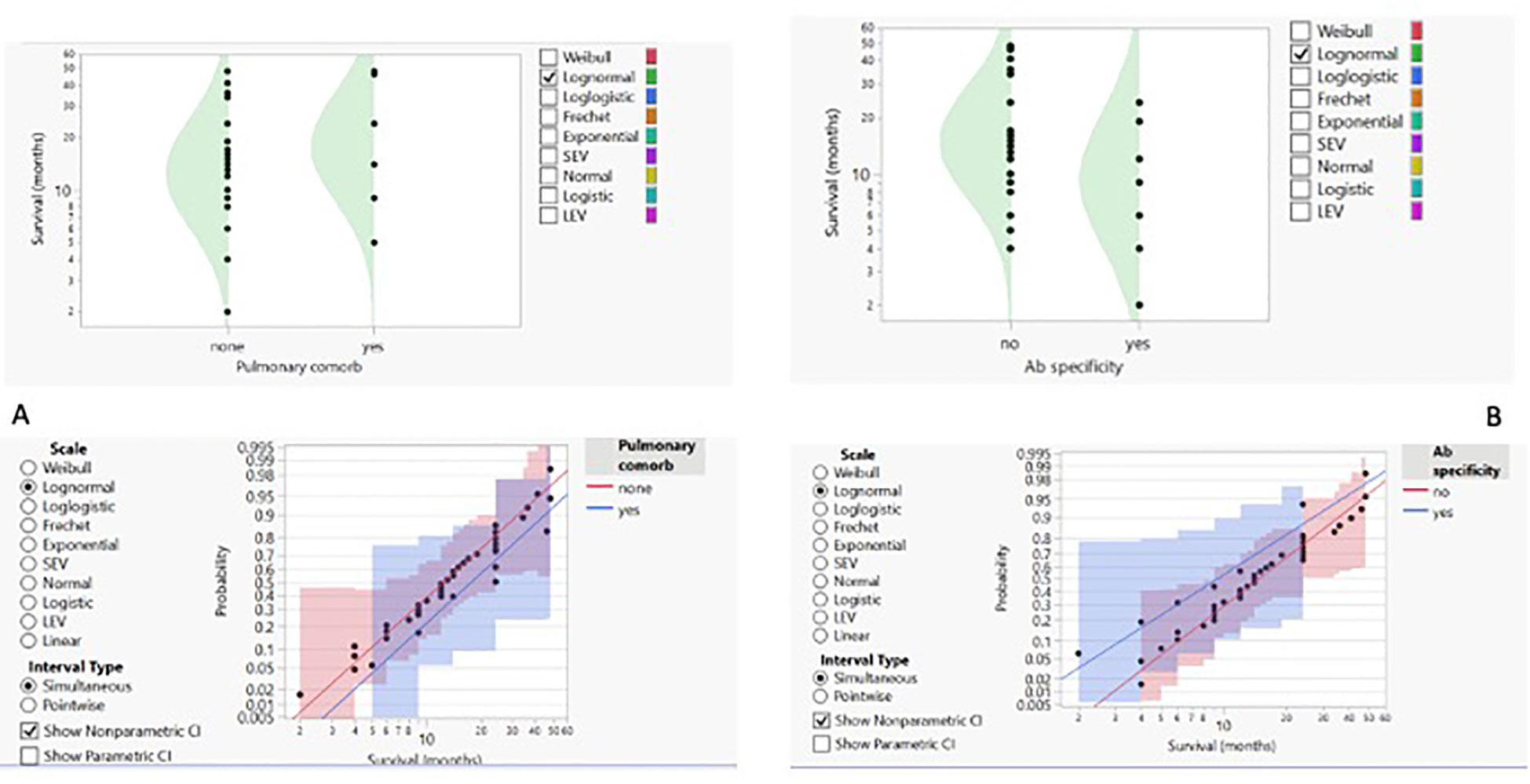
Figure 3. (A) Concomitant pulmonary comorbidity (COPD) and probability of survival shown as density curves with logarithmic fit–nonparametric overlay. (B) Detection of autoantibodies and probability of survival presented as density curves with logarithmic fit–nonparametric overlay.
3.3 Integrating molecular profiles into the clinical frameworkWith respect to IPF-associated cancer cases, we then proceed with a descriptive analysis not connected to outcomes and evaluate their molecular features to expand the knowledge base and better characterise malignant onset and progression. Recent evidence suggests a profibrotic role of the PD1/PD-L1 axis in IPF and mediastinal lymph nodes (30–32), paving the way for the therapeutic exploitation of low doses of immune checkpoint inhibitor (33, 34). These data strongly support the rationale for evaluating the expression of PD-L1 in IPF-associated cancer (Figures 4A, B). With respect to the cohort analysed, a key difference in PD-L1 tumour expression is related to patients’ gender, with significantly higher expression in women. Notably, this finding is not related to survival nor to autoantibody detection, and—quite unexpectedly—PD-L1 expression is uniformly distributed among current, past, and never smokers. We then investigated the impact of EGFR status and found that the wild-type receptor was associated with the highest PD-L1 levels (Figure 4C) and the presence of autoantibodies (Figure 4D). Moreover, when applied, the partition analysis for EGFR status (Figure 4E) confirmed that the wild-type status was more frequently detected in male and smoker subjects.
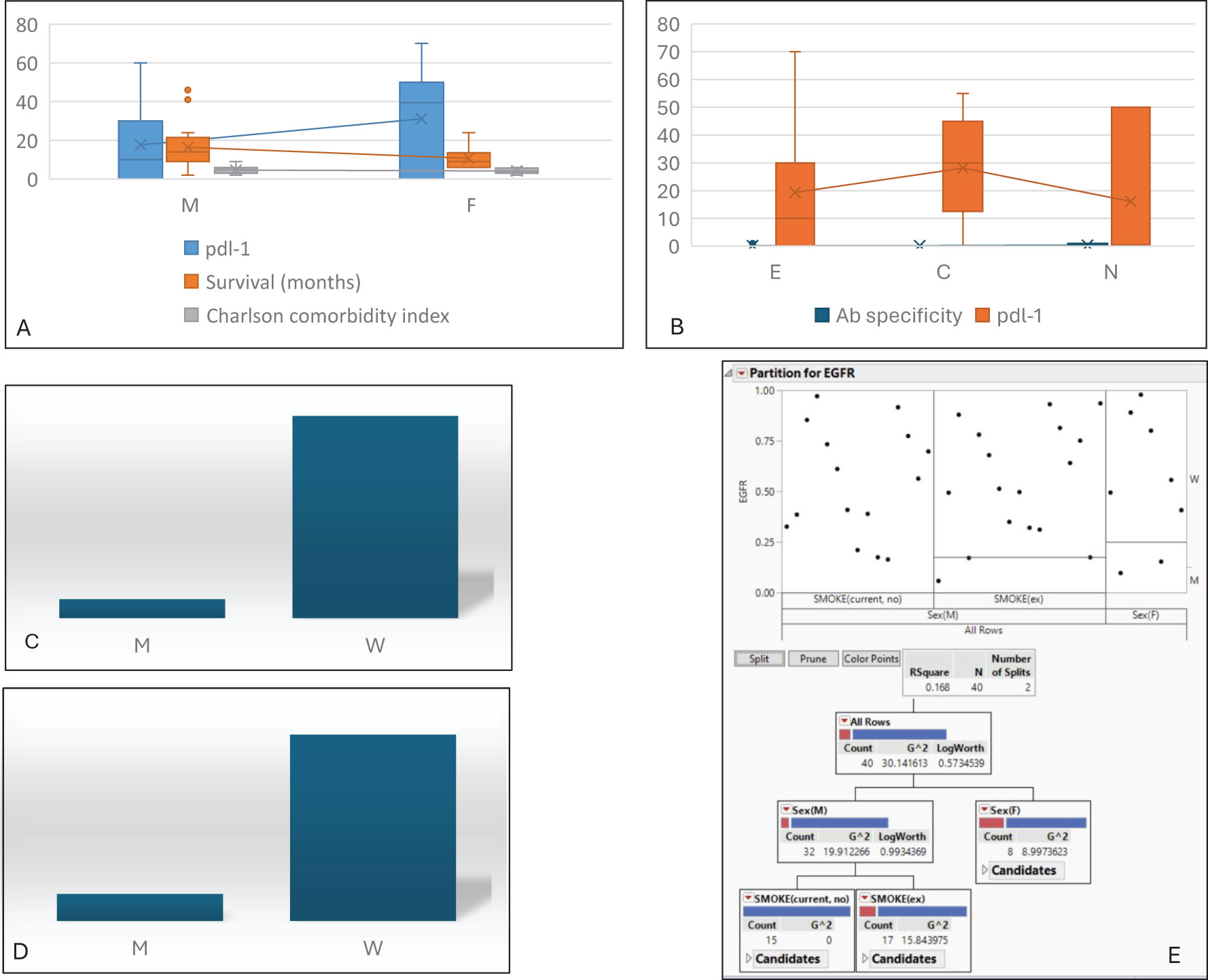
Figure 4. Molecular asset. (A) Distribution of PDL-1 level, survival, and CMI based on gender differences. (B) Distribution of autoantibody detection and PDL-1 levels based on smoking habits. Analysis of EGFR status vs. total amount of PDL-1 (C) and autoantibody detection (D). (E) Partition analysis for EGFR status vs. gender and smoking, confirming that the wild-type receptor is more frequently associated with male sex and smoking habits. M, male; F, female; CMI, Charlson Comorbidity Index; E, past smoker; C, current smoker; N, never smoker; M, mutated; W, wild type.
4 DiscussionThe present study focuses on a retrospective analysis of a dataset regarding clinicopathologic and laboratory features of LC associated with ILD, collected from three reference centres in Italy. The design of the study itself limits mechanistic conclusions from the analyses; however, the statical approach used allows for the identification of previously unsuspected relationships between variables, highlighting critical pattern matches that could warrant future perspectives and experimental works. Thus, within this limitation, some considerations should be noted. The first is that, among ILDs, a selective preference for malignant growth in the IPF-related milieu can be recognised (35). Moreover, patients’ gender acts as an independent factor associated with overall survival. This trend seems to contrast with previously published data reporting no gender differences in survival in ILD patients with cancer (36), although a proper comparison between different studies should be limited by case heterogeneity. The significant difference in terms of lower survival in women with IPF and LC is an unexpected finding, not reported before in the literature. Men with IPF and LC, if subjected to surgery but in the absence of autoantibody detection, have a better prognosis. It is known that women have a greater predisposition to autoimmune disease (37–39), and this fact, in association with lower rates of surgical treatments compared to those in men (χ2: 5.17, p-value: 0.05 degrees of freedom), may be in some way related to lower survival. This observation allows us to conclude that, while being a woman with IPF is not associated with better survival, as already reported (40), women with concurrent IPF and LC had worse survival compared to male patients. Although limited, this result clearly suggests that surgical excision should be considered an effective option in early-stage LC in fibrotic patients—just as in nonfibrotic ones—and seems to be superior to radiotherapy in comparable populations. With respect to the cohort analysed, it should be underlined that women were rarely selected for surgery, and this issue should have had an impact on their survival. No clinical reasons emerged as causatively related to this observation, and these data support a rationale for a deeper investigation into the sociocultural role played by gender in the management of rare conditions such as IPF combined with LC. Interestingly, all except two patients who underwent surgery were treated with pirfenidone, which has already been reported to be associated with a lower risk of postoperative exacerbation (41, 42). Although the detection of autoantibodies in interstitial pneumonia with autoimmune features (IPAF) has been reported to be associated with better outcomes (43), more recent observations suggest that their presence in IPF should be associated with more rapid disease progression (44, 45). On the other hand, it should be noted that in scleroderma, the occurrence of specific autoantibodies (including anti-Scl70) has been strictly associated with a higher risk of cancer onset (46, 47). Thus, although autoantibody in IPF still requires full clarification, their detection in IPF associated with LC indicates poorer patient outcomes with potential implications. To further validate the study hypothesis, which is limited by the retrospective nature of the analysed data, we aim to produce exploratory findings that, after prospective validation, could help in the further development of a personalised approach to IPF-related LC. The results of this analysis highlight a gender-related expression of PD-L1 that is largely independent of smoking habits and more frequently associated with EGFR wild-type status. This observation allows us to hypothesise that immune checkpoint expression in this cancer type may be related to its activation in the surrounding fibrotic stroma rather than in response to cigarette smoke, as reported in primary lung cancer (48, 49). Thus, although genetic-driven tumour profiling is similar to that of IPF-free LC, as documented by EGFR behaviour and asset, the immune tumour status should be strictly related to the surrounding IPF stroma and the clinicopathologic features of the interstitial diseases. This preliminary observation suggests a strong rationale for further experimental research aimed at defining whether IPF-associated LC, once initiated, exploits the surrounding IPF traits as a selective force for progression.
5 ConclusionsAlthough limited, this study suggests that IPF is, among the ILDs, the one most strongly associated with LC onset.
Interestingly, our data are consistent with already published studies showing that autoantibodies are associated with worse IPF progression, according to mechanisms that involve the potential role of autoreactive B cells (see, for instance, refs (43, 44, 50).).
Although the role of autoantibodies in the pathogenesis of CTD-ILDs is better defined, recent data in the literature suggest an active role of B cells, plasma cells, and autoantibodies in IPF as well (51, 52). For example, in the study by Koether et al. (44), IPF patients exhibit an altered B-cell phenotype, and those with autoantibodies against the protein periplakin experience a rapid decline of lung function, suggesting a role for them in the progression of the disease. Tertiary lymphoid structures (sites of onset of humoral responses) have been found in the biopsies and explanted lungs of IPF patients, with a higher rate of activation of cells (assessed by the expression of CD40) in the advanced stages of the disease (53). The presence of prominent B-cell activation may contribute to the fibrotic remodeling process and suggest a potential role for anti-CD20 monoclonal antibodies in the treatment of IPF, similar to non-IPF ILDs. Both innate and adaptative immunity contribute to the fibrotic response characterising IPF. Although the role of T cells is better recognised in autoimmune ILDs or antigen-driven ILDs, growing evidence shows a role of T helper cells in IPF. Experimental models suggest a prominent role of Th2 and Th 17 subsets and their cytokines (54). In the context of excessive activation of T cells, a role may be played by a dysregulation of PD-1/PDL-1 axis, which has an inhibitory role on T-cell response. For example, in one study, PD-1 expression was found significantly downregulated in the T helper cells derived from the BAL of 20 patients with several ILDs, including IPF. This finding may explain the increased T-cell activity detected in these patients (55). Similarly, a population of T helper cells from peripheral blood mononuclear cells, with high expression of PD-1 and a cytotoxic phenotype, has been identified by mass cytometry in a population of SSc patients and has been associated with the presence and the severity of ILD. Overall, although of relevant clinical interest, our results cannot allow mechanistic conclusions/implications. Being a retrospective study, the total antibody levels could not be evaluated, nor could B-cell activation and soluble levels of PDL1. The findings of our study give a strong rationale for the development of perspective studies focusing on this topic, with potential gender-associated basis. Moreover, the results of the present work allow us to conclude that IPF-associated lung cancer can be defined as a separate malignant entity, carrying a definite clinicopathologic profile. This issue raises a strong rationale for the design of further studies aimed at evaluating the molecular asset of this cancer type. A second point that deserves deeper consideration is that, being a rare condition, IPF-associated LC should be managed by a multidisciplinary board with specific expertise and know-how in referral centers, as these patients could benefit, in some instances, from active and even aggressive (surgical) treatments.
Data availability statementThe datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/supplementary material.
Ethics statementThe study entered a main project that was approved by local Ethical Commission and each enrolled patient gave written informed consent before enrolment (Comitato di Bioetica, Fondazione IRCCS Policlinico San Matteo, approval numbers: protocol #20090002344; procedure # 20090019080; date of approval: June 3rd, 2009). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and institutional requirements.
Author contributionsGMS: Conceptualization, Data curation, Investigation, Software, Writing – original draft, Writing – review & editing. FP: Conceptualization, Investigation, Methodology, Software, Writing – original draft, Writing – review & editing. DL: Data curation, Investigation, Methodology, Software, Writing – original draft, Writing – review & editing. VD’A: Conceptualization, Data curation, Investigation, Methodology, Writing – original draft. AB: Methodology, Supervision, Writing – original draft, Writing – review & editing. GS: Investigation, Methodology, Writing – original draft. PT: Data curation, Investigation, Methodology, Supervision, Writing – original draft. MFB: Investigation, Methodology, Writing – original draft, Writing – review & editing. FM: Data curation, Investigation, Methodology, Writing – original draft. SL: Data curation, Investigation, Methodology, Software, Writing – original draft. LFD: Data curation, Investigation, Methodology, Writing – original draft. SM: Conceptualization, Investigation, Writing – original draft. DP: Conceptualization, Investigation, Writing – original draft. TO: Investigation, Methodology, Writing – original draft. CB: Data curation, Investigation, Methodology, Writing – original draft. AG: Formal analysis, Supervision, Writing – original draft. LC: Conceptualization, Data curation, Writing – review & editing. IC: Conceptualization, Data curation, Writing – review & editing.
FundingThe author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was funded by Ricerca corrente 5x1000-2020 (cod. 090000X121-prot. 08050122) to GS.
AcknowledgmentsAndrea PG Marchelli is acknowledged for support and revision of the statistical analysis, and Andrew Raneri III for revision of the English language. GMS would like to thank FTR for insightful discussions and enthusiastic support.
Conflict of interestThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
AbbreviationsILD, interstitial lung diseases; IPF, idiopathic pulmonary fibrosis; NSIP, nonspecific interstitial pneumonia; COPD, chronic obstructive pulmonary disease; LC, lung cancer; SCLC, small cell lung cancer; NSCLC, non-small cell lung cancer; SCC, squamous cell carcinoma; ADC, adenocarcinoma; PM, pleural mesothelioma; BMI, body mass index; ECOG, Eastern Cooperative Oncology Group; PD-L1: programmed-death ligand 1; TPS, tumour proportion score; EGFR, epidermal growth factor receptor; CCI, Charlson Comorbidity Index; PH, pulmonary hypertension; GERD, gastroesophageal reflux disease; BPH, benign prostatic hyperplasia; CKD, chronic kidney disease; P, pirfenidone; N, nintedanib; MTX, methotrexate; Cts, corticosteroids.
References1. Shah Gupta R, Koteci A, Morgan A, George PM, Quint JK. Incidence and prevalence of interstitial lung diseases worldwide: a systematic literature review. BMJ Open Respir Res. (2023) 10:e001291. doi: 10.1136/bmjresp-2022-001291
PubMed Abstract | Crossref Full Text | Google Scholar
2. Brixey AG, Oh AS, Alsamarraie A, Chung JH. Pictorial review of fibrotic interstitial lung disease on high resolution CT scan and updated classification. Chest. (2024) 165:908–23. doi: 10.1016/j.chest.2023.11.037
PubMed Abstract | Crossref Full Text | Google Scholar
3. Amati F, Spagnolo P, Ryerson CJ, Oldham JM, Gramegna A, Stainer A, et al. Walking the path of treatable traits in interstitial lung diseases. Respir Res. (2023) 24:251. doi: 10.1186/s12931-023-02554-8
PubMed Abstract | Crossref Full Text | Google Scholar
5. Hutchinson J, Fogarty A, Hubbard R, McKeever T. Global incidence and mortality of idiopathic pulmonary fibrosis: a systematic review. Eur Respir J. (2015) 46:795–806. doi: 10.1183/09031936.00185114
PubMed Abstract | Crossref Full Text | Google Scholar
7. Lee JH, Park HJ, Kim S, Kim YJ, Kim HC. Epidemiology and comorbidities in idiopathic pulmonary fibrosis: a nationwide cohort study. BMC Pulm Med. (2023) 23:54. doi: 10.1186/s12890-023-02340-8
PubMed Abstract | Crossref Full Text | Google Scholar
8. Vancheri C, Failla M, Crimi N, Raghu G. Idiopathic pulmonary fibrosis: a disease with similarities and links to cancer biology. Eur Respir J. (2010) 35:496–504. doi: 10.1183/09031936.00077309
PubMed Abstract | Crossref Full Text | Google Scholar
9. Stella GM, Inghilleri S, Pignochino Y, Zorzetto M, Oggionni T, Morbini P, et al. Activation of oncogenic pathways in idiopathic pulmonary fibrosis. Transl Oncol. (2014) 7:650–5. doi: 10.1016/j.tranon.2014.05.002
PubMed Abstract | Crossref Full Text | Google Scholar
10. Selman M, Pardo A. Idiopathic pulmonary fibrosis: from common microscopy to single-cell biology and precision medicine. Am J Respir Crit Care Med. (2024) 209:1074–81. doi: 10.1164/rccm.202309-1573PP
PubMed Abstract | Crossref Full Text | Google Scholar
11. Kewalramani N, Machahua C, Poletti V, Cadranel J, Wells AU, Funke-Chambour M. Lung cancer in patients with fibrosing interstitial lung diseases: an overview of current knowledge and challenges. ERJ Open Res. (2022) 8:00115–2022. doi: 10.1183/23120541.00115-2022
PubMed Abstract | Crossref Full Text | Google Scholar
12. Karampitsakos T, Spagnolo P, Mogulkoc N, Wuyts WA, Tomassetti S, Bendstrup E, et al. Lung cancer in patients with idiopathic pulmonary fibrosis: A retrospective multicentre study in Europe. Respirology. (2023) 28:56–65. doi: 10.1111/resp.14363
PubMed Abstract | Crossref Full Text | Google Scholar
13. Perrotta F, Chino V, Allocca V, D'Agnano V, Bortolotto C, Bianco A, et al. Idiopathic pulmonary fibrosis and lung cancer: targeting the complexity of the pharmacological interconnection. Expert Rev Respir Med. (2022) 16:1043–55. doi: 10.1080/17476348.2022.2145948
PubMed Abstract | Crossref Full Text | Google Scholar
14. Nicholson AG, Tsao MS, Beasley MB, Borczuk AC, Brambilla E, Cooper WA, et al. The 2021 WHO classification of lung tumors: impact of advances since 2015. J Thorac Oncol. (2022) 17:362 387. doi: 10.1016/j.jtho.2021.11.003
PubMed Abstract | Crossref Full Text | Google Scholar
16. Stella GM, Senetta R, Cassenti A, Ronco M, Cassoni P. Cancers of unknown primary origin: current perspectives and future therapeutic strategies. J Transl Med. (2012) 10:12. doi: 10.1186/1479-5876-10-12
PubMed Abstract | Crossref Full Text | Google Scholar
17. Singh N, Temin S, Baker S Jr, Blanchard E, Brahmer JR, Celano P, et al. Therapy for stage IV non-small-cell lung cancer without driver alterations: ASCO living guideline. J Clin Oncol. (2022) 40:3323–43. doi: 10.1200/JCO.22.00825
PubMed Abstract | Crossref Full Text | Google Scholar
18. Lie M, Khambata-Ford S, Copie-Bergman C, Huang L, Juco J, Hofman V, et al. Use of the 22C3 anti-PD-L1 antibody to determine PD-L1 expression in multiple automated immunohistochemistry platforms. PLoS One. (2017) 12:e0183023. doi: 10.1371/journal.pone.0183023
PubMed Abstract | Crossref Full Text | Google Scholar
19. Kaira K, Yamaguchi O, Kawasaki T, Hashimoto K, Miura Y, Shiono A, et al. Prognostic significance of tumor infiltrating lymphocytes on first-line pembrolizumab efficacy in advanced non-small cell lung cancer. Discovery Oncol. (2023) 14:6. doi: 10.1007/s12672-023-00615-4
PubMed Abstract | Crossref Full Text | Google Scholar
20. Pan B, Kang Y, Jin Y, Yang L, Zheng Y, Cui L, et al. Automated tumor proportion scoring for PD-L1 expression based on multistage ensemble strategy in non-small cell lung cancer. J Transl Med. (2021) 19:249. doi: 10.1186/s12967-021-02898-z
PubMed Abstract | Crossref Full Text | Google Scholar
21. Liu J, Zheng Q, Mu X, Zuo Y, Xu B, Jin Y, et al. Automated tumor proportion score analysis for PD-L1 (22C3) expression in lung squamous cell carcinoma. Sci Rep. (2021) 11:15907. doi: 10.1038/s41598-021-95372-1
PubMed Abstract | Crossref Full Text | Google Scholar
22. Jing C, Mao X, Wang Z, Sun K, Ma R, Wu J, et al. Next-generation sequencing-based detection of EGFR, KRAS, BRAF, NRAS, PIK3CA, Her-2 and TP53 mutations in patients with non-small cell lung cancer. Mol Med Rep. (2018) 18:2191–7. doi: 10.3892/mmr.2018.9210
PubMed Abstract | Crossref Full Text | Google Scholar
23. de Biase D, Visani M, Malapelle U, Simonato F, Cesari V, Bellevicine C, et al. Next-generation sequencing of lung cancer EGFR exons 18-21 allows effective molecular diagnosis of small routine samples (cytology and biopsy). PLoS One. (2013) 8:e83607. doi: 10.1371/journal.pone.0083607
PubMed Abstract | Crossref Full Text | Google Scholar
24. Kang DW, Park SK, Kang S, Lee EK. Cost-effectiveness of next-generation sequencing for advanced EGFR/ALK-negative non-small cell lung cancer. Lung Cancer. (2024) 197:107970. doi: 10.1016/j.lungcan.2024.107970
PubMed Abstract | Crossref Full Text | Google Scholar
25. Podolanczuk AJ, Thomson CC, Remy-Jardin M, Richeldi L, Martinez FJ, Kolb M, et al. Idiopathic pulmonary fibrosis: state of the art for 2023. Eur Respir J. (2023) 61:2200957. doi: 10.1183/13993003.00957-2022
PubMed Abstract | Crossref Full Text | Google Scholar
26. Mai TH, Han LW, Hsu JC, Kamath N, Pan L. Idiopathic pulmonary fibrosis therapy development: a clinical pharmacology perspective. Ther Adv Respir Dis. (2023) 17:17534666231181537. doi: 10.1177/17534666231181537
PubMed Abstract | Crossref Full Text | Google Scholar
27. Raghu G, Remy-Jardin M, Richeldi L, Thomson CC, Inoue Y, Johkoh T, et al. Idiopathic pulmonary fibrosis (an update) and progressive pulmonary fibrosis in adults: an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. (2022) 205:e18–47. doi: 10.1164/rccm.202202-0399ST
PubMed Abstract | Crossref Full Text | Google Scholar
28. Kuwana M, Gil-Vila A, Selva-O'Callaghan A. Role of autoantibodies in the diagnosis and prognosis of interstitial lung disease in autoimmune rheumatic disorders. Ther Adv Musculoskelet Dis. (2021) 13:1759720X211032457. doi: 10.1177/1759720X211032457
PubMed Abstract | Crossref Full Text | Google Scholar
29. Stevenson BR, Thompson GA, Watson MC, Bundell CS, Klinken EM, John M, et al. Autoantibodies in interstitial lung diseases. Pathology. (2019) 51:518–52. doi: 10.1016/j.pathol.2019.03.007
PubMed Abstract | Crossref Full Text | Google Scholar
31. Yang L, Xia H, Gilbertsen A, Smith K, Racila E, Bitterman PB, et al. IL-8 concurrently promotes idiopathic pulmonary fibrosis mesenchymal progenitor cell senescence and PD-L1 expression enabling escape from immune cell surveillance. Am J Physiol Lung Cell Mol Physiol. (2023) 324:L849–62. doi: 10.1152/ajplung.00028.2023
PubMed Abstract | Crossref Full Text | Google Scholar
32. Jiang A, Liu N, Wang J, Zheng X, Ren M, Zhang W, et al. The role of PD-1/PD-L1 axis in idiopathic pulmonary fibrosis: Friend or foe? Front Immunol. (2022) 13:1022228. doi: 10.3389/fimmu.2022.1022228
留言 (0)