Probiotic strains and culture conditions
The probiotics used in this research work (Table 1) were acquired from the JHJ Company (Nowa Wies, Gizalki, Poland). Before use, the probiotic cultures were kept at − 80 °C in 50% glycerol. All strains were cultured and maintained in MRS broth at 37 °C. A total of 6.82 g of MRS agar (de Man, Rogosa and Sharpe, Merck KGaA, Darmstadt) was dissolved in 100 mL of distilled water and stirred (IKA® RCT basic IKAMAG™ Safety Control Magnetic Stirrer) to dissolve the agar completely. Next, it was autoclaved at 121 °C for 15 min and vortexed for 10 s. Stock cultures of probiotic strains were established on agar plates, and the plates underwent a 24-h incubation period at 37 °C. A bacterial suspension was prepared for each strain in 10 mL of DeMan, Rogosa, and Sharpe broth (MRS) (Merck KGaA, Darmstadt). Subsequently, a 96-well microplate was filled with 250 µL of MRS broth and 10 µL of each bacterial suspension. The microplate was then incubated for 48 h at 37 °C under aerobic conditions. Three replicates of each sample were included for each of the three repetitions for each probiotic. MRS broth without inoculum was used as a control. Bacterial growth measurements (OD600) were performed every 12 h using a Multiskan™ FC Microplate Photometer and SkanIt software version 7.0 (Thermo Fisher Scientific, Waltham, MA). Microtiter plates were shaken for 10 s before the microplate readings were taken to ensure homogeneity in the samples.
Table 1 Concentrations of probiotics used for the 2,2-diphenyl-1-picrylhydrazyl (DPPH) DPPH testIn vitro determination of the antioxidant activities of the selected probiotics
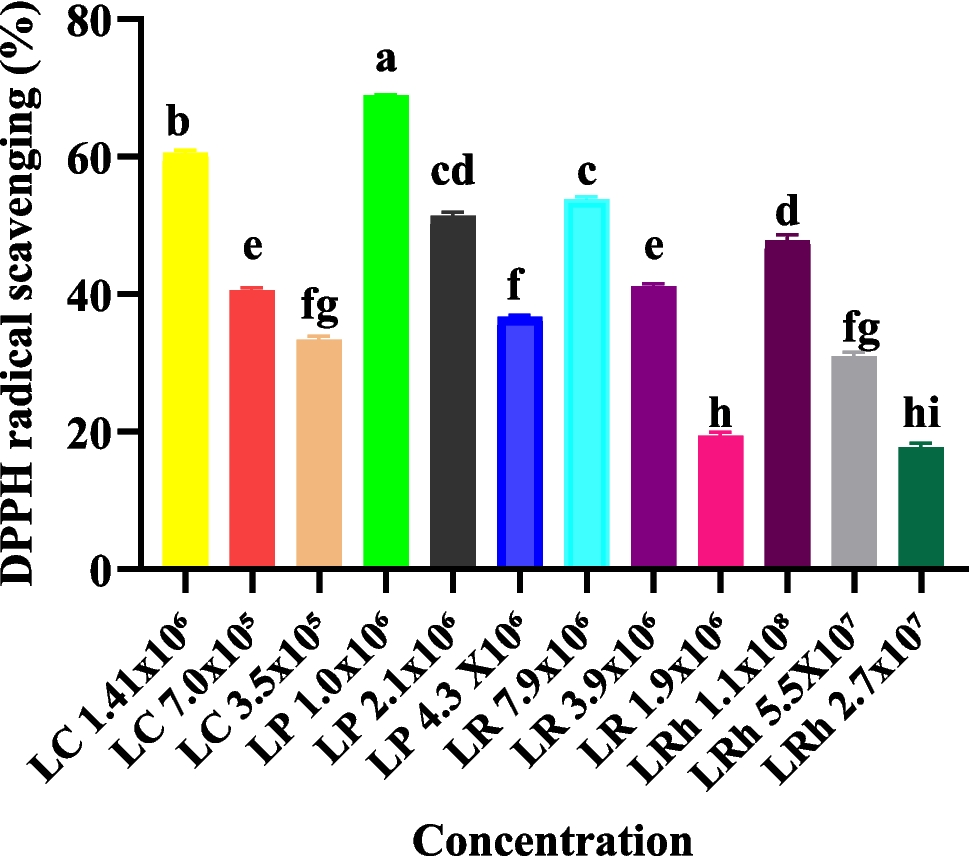
The list of Lactobacillus strains used in this study is provided in Table 1. To pre-select probiotics for in ovo injection, we used the 2,2-diphenyl-1-picrylhydrazyl (DPPH) (Sigma‒Aldrich, St. Louis, MO) assay to measure free radical scavenging activities according to (Kao and Chen 2006).
Briefly, 0.1 mM of DPPH was dissolved in 100 mL of ethanol. The mixture was vigorously shaken and left to react for 30 min at room temperature in the dark. It was always used fresh on the day of analysis. Prior to the DPPH assay, the Lactobacillus samples were serially diluted, and 10 µl of sample (with appropriate dilution), 190 µl of sample, and the control (200 µl of DPPH ethanolic solution) were added to a 96-well microtiter plate. The blank group contained MRS broth media and ethanol. The optical absorbance at 520 nm was measured in triplicate using a MultiskanTM FC Microplate Photometer. Using the following equation below, the percentage of free radical scavenging activity was determined.
$$\left(\%\right) \text = [(\text-\text)/\text] \times 100$$
where Ac is the absorbance of the control and As is the absorbance of the sample.
The results are expressed as the EC50 (μg/mL), which is the lowest antioxidant concentration needed to reduce 50% of the initial DPPH reaction from the moment the extract reached stability. Based on the growth curve and the DPPH assay results, the bioactive compound with the best growth and highest antioxidant activity was selected for in ovo application to validate its effects on Ross 308 broiler chickens. The prebiotic GOS was selected for in ovo application studies based on results of previous studies by our group showing its ability to mitigate heat stress in Ross 308 broilers (Slawinska et al. 2020b).
Egg incubation and in ovo protocol
A total of three hundred (300) fertile Ross 308 broiler eggs were incubated under standard incubation conditions (Midi series I, Fest Incubators, Gostyń, Poland). On day 7 of embryonic development, eggs were taken out of the incubator and sterilized using 70% ethanol, then candled, and the infertile and dead embryos were discarded. The remaining fertile eggs were randomly allotted into four groups: negative control (NC), positive control (PC), GOS, and LP. Next, a 20G needle was used to make a hole in the air chamber of the eggs. Subsequently, in ovo injection was manually performed on the 12th day of egg incubation in all the groups except the NC group. A 0.2 mL sterile 0.9% physiological saline solution was injected into the PC group eggs while the GOS group eggs were injected with 3.5 mg of GOS/egg suspended in 0.2 mL of physiological saline and the LP group was injected with 106 CFU of LP bacteria/egg suspended in 0.2 mL of physiological saline solution. After injection, each egg was sealed using organic glue (Elmer’s school glue, Elmer’s Products Inc., USA), and immediately returned to the incubator.
Birds and housing
The experiment was conducted in compliance with the Ethics Committee for Experiments with Animals guidelines and the Polish Act on the Protection of Animals Used for Scientific or Educational Purposes regulations of January 15, 2015 (which was implemented by the European Parliament and Council of September 22, 2010, Directive 2010/63/EU on the protection of animals used for scientific purposes). All birds in each experimental group consisting of 32 birds/pens were housed in separate pens with similar optimized environmental conditions during the experiment. Water and feed were made available to the chickens at all times. The birds were fed the following three types of age-dependent diets throughout the experimental period: starter (1–21 days), grower (22–28 days), and finisher (29–35 days), consisting of 12.45, 13.01, and 13.01 MJ/kg of metabolizable energy and 22.3%, 20.2%, and 20.2% crude protein, respectively. The dietary mixtures were in accordance with broiler chicken dietary requirements (Smulikowska and Rutkowski, 2018). The initial environmental temperature in the pens was 32–33 °C on day one of life, and the temperature steadily reduced reaching approximately 21 °C at the end of the trial period.
Sample collection
At the end of the rearing period, 8 birds per group (n = 24) with a final average body weight of 2.43–2.53 kg were randomly chosen. The birds were slaughtered by decapitation after being deprived of feed for 10 h and left to bleed for approximately 90 s. Following the slaughtering of each bird, two milliliters of blood were collected in K-EDTA tubes and centrifuged at 3000 × g for 15 min to extract plasma. Next, the plasma samples were immediately placed on dry ice and transported to the laboratory. Upon arrival, all the samples were kept at − 80 °C until analysis. In addition, cecal mucosa, liver, spleen, and breast muscle were collected and preserved in RNA stabilization reagent (fix RNA: E0280, EURx, Gdańsk, Poland) and transported at room temperature, and the fixed RNA was poured off and the tubes with the samples were kept at − 80 °C until use.
RNA extraction, RT‒PCR, and qPCR gene expression analysis
Tissues were homogenized with a TissueRuptor homogenizer (990,890, Qiagen, Wrocław, Poland) and immersed in a tube containing 1 mL of RNA extracol solution (E3700, EURx, Gdańsk, Poland) for the RNA isolation procedure. Next, each sample was centrifuged using 0.2 mL of chloroform (112,344,305, Chempur, Piekary Śląskie, Poland). A commercial kit (Universal RNA purification kit (E3598, EURx, Gdańsk, Poland)) was used to carry out the subsequent steps of the RNA isolation process. A NanoDrop 2000 spectrophotometer (Thermo Scientific, Warsaw, Poland) was used to measure the quantity and quality of the RNA, while a 2% agarose gel was used to assess RNA integrity. RNA samples were stored at − 80 °C until use. Using the smART First Strand cDNA Synthesis Kit (0804, EURx, Poland), the RT-PCR process was performed following the manufacturer’s protocol. Next, the cDNA obtained was diluted to 100 ng/μl. Afterward, RT‒qPCR was carried out using a total volume of 10 μL. The reaction mixture included Maxima SYBR Green qPCR Master Mix (0401, EURx, Gdańsk, Poland), 1 μM of each primer, and 2 μl of diluted cDNA. Thermal cycling was conducted using a LightCycler II 480 (Roche Diagnostics, Basel, Switzerland). Each RT‒qPCR was carried out in two technical replicates in 96-well plates (4TI-0955, AZENTA, Genomed, Warszawa, Poland). The qPCR protocol for the gene expression analysis consisted of initial denaturation for 15 min (95 °C), followed by 40 cycles of amplification (denaturation at 95 °C for 15 s, annealing at 58 °C for 30 s, and elongation at 72 °C for 30 s).
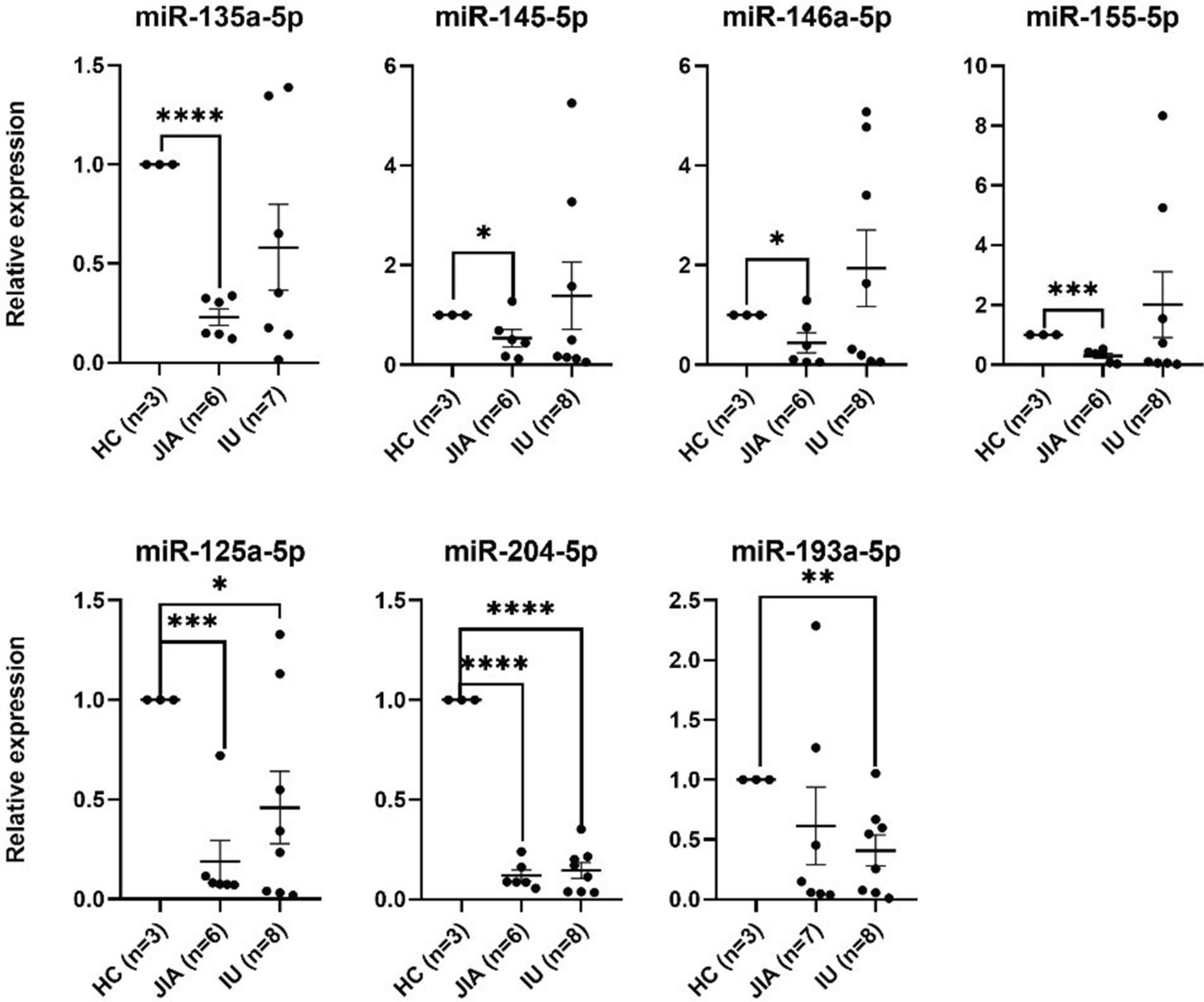
The expression levels of the target genes were determined via geometric means of two housekeeping genes (Actb and G6pdh). The target genes analyzed for each tissue and the reference genes are listed in Table 2. The relative gene expression was calculated using the ΔΔCt method. The ΔCt of the control group was subtracted from the ΔCt of each of the treatment groups. The fold change (FC) of the target gene in the treatment group against the control group was calculated as 2−∆∆Ct.
Table 2 List of target genes used for qPCR gene expression analysisBlood plasma metabolite analysis
Blood plasma from eight 35 d old birds per each experimental group was randomly chosen to analyze metabolite concentrations and enzyme activities. An automatic enzyme analyzer (Pentra C 400, Axon Lab AG, Germany) was used to determine aspartate aminotransferase (AST): Kit No. A11A01629; alanine aminotransferase (ALT): A11A01627; high-density lipoprotein (HDL): A11A01636; low-density lipoprotein (LDL): A11A01638; total cholesterol: Kit No. A11A01634; triglyceride (TG): Kit No. A11A01640 (Horiba ABX), non-esterified fatty acid (NEFA): Kit No. 434–91,795 (Wako Chemicals GmbH, Neuss, Germany)); uric acid: Kit No. A11A01670; glucose: Kit No. A11A01667; lactose dehydrogenase (LDH): Kit No. A11A01871; and gamma-glutamyl transferase (GGT): Kit No. A11A01630 (Axon Lab AG, Reichenbach, Germany). These selected plasma metabolite parameters were analyzed at the Institute of Nutritional Physiology at the Research Institute for Farm Animal Biology (FBN), Dummerstorf, Germany.
Statistical analysis
All the data were checked for distribution normality, presence of outliers, and homogeneity of variance using the Shapiro–Wilk test and Levene test, respectively. The DPPH in vitro results and plasma metabolites were analyzed using GraphPad Prism version 10.1.2. The one-way ANOVA was used for the DPPH in vitro data. To analyze the plasma metabolites, we used principal component analysis (PCA). Tukey’s HSD test was used to determine the differences between means (P < 0.05). The ΔCt values of every treatment group were compared with those of the control group for the gene expression analysis using GraphPad Prism and Student’s t-test to identify significant differences among the treatments (P < 0.05), and plotting of graphs was done with Microsoft Excel.




留言 (0)