
記住我
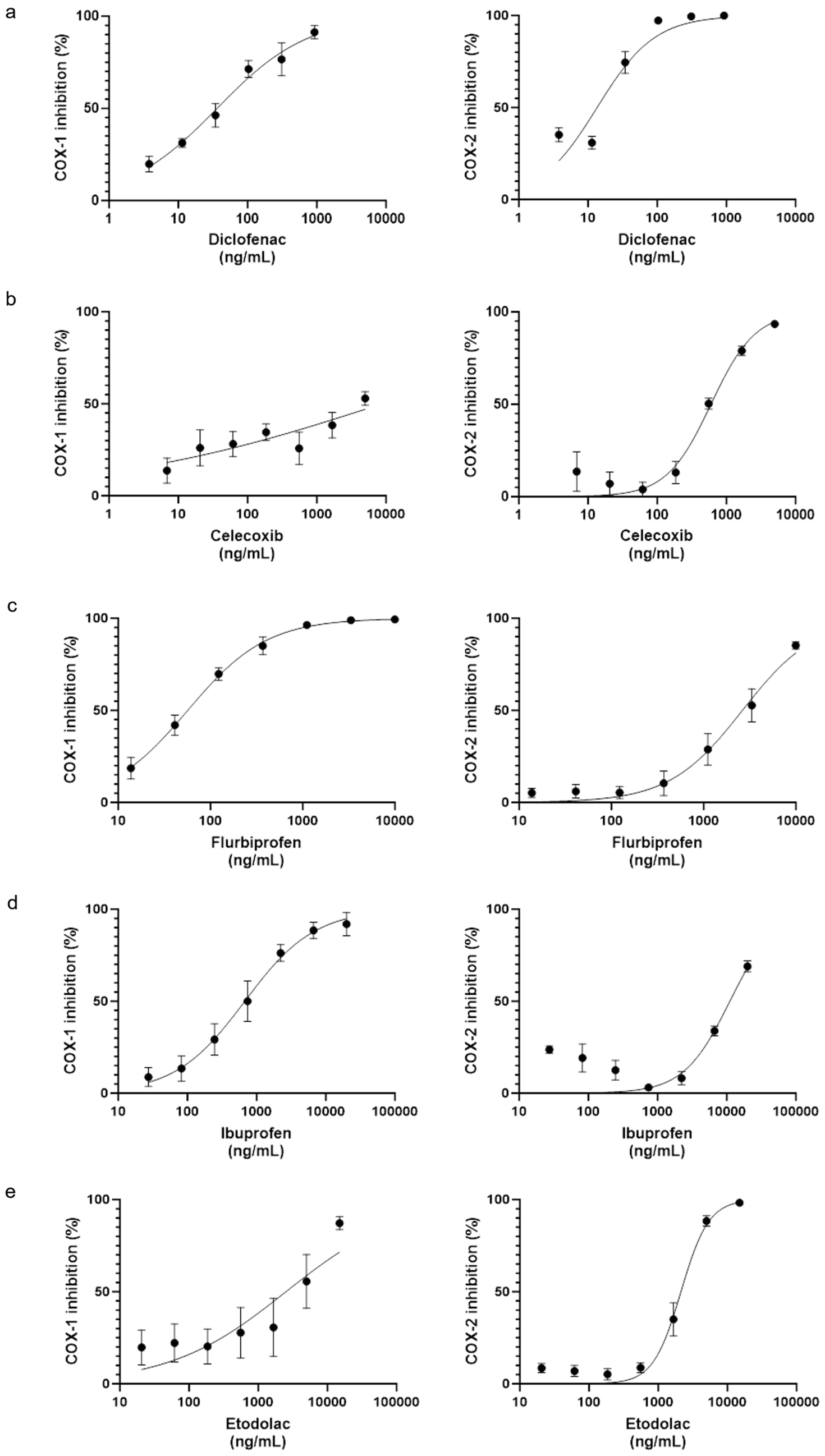

The primary objective of this study is to assess the effectiveness of ultrasound-guided thoracic paravertebral PRP injections in alleviating acute ZAP and preventing the onset of PHN. The secondary objective is to compare the outcomes of PRP injections with traditional antiviral therapy in treating acute HZ.
Study Design and RecruitmentThis study is a prospective, randomized, controlled, open-label, endpoint-blinded single-center trial. We plan to recruit 128 patients with ZAP at the Second Affiliated Hospital of Guangxi Medical University. Before the start of the study, all researchers will receive standardized training, including trial content, treatment strategies, treatment methods, evaluation and quality control, etc., and pass the assessment. Patients will be randomly assigned to the thoracic paravertebral PRP injection group or the antiviral therapy group at a 1:1 ratio. Recruitment will be conducted by experienced attending physicians.
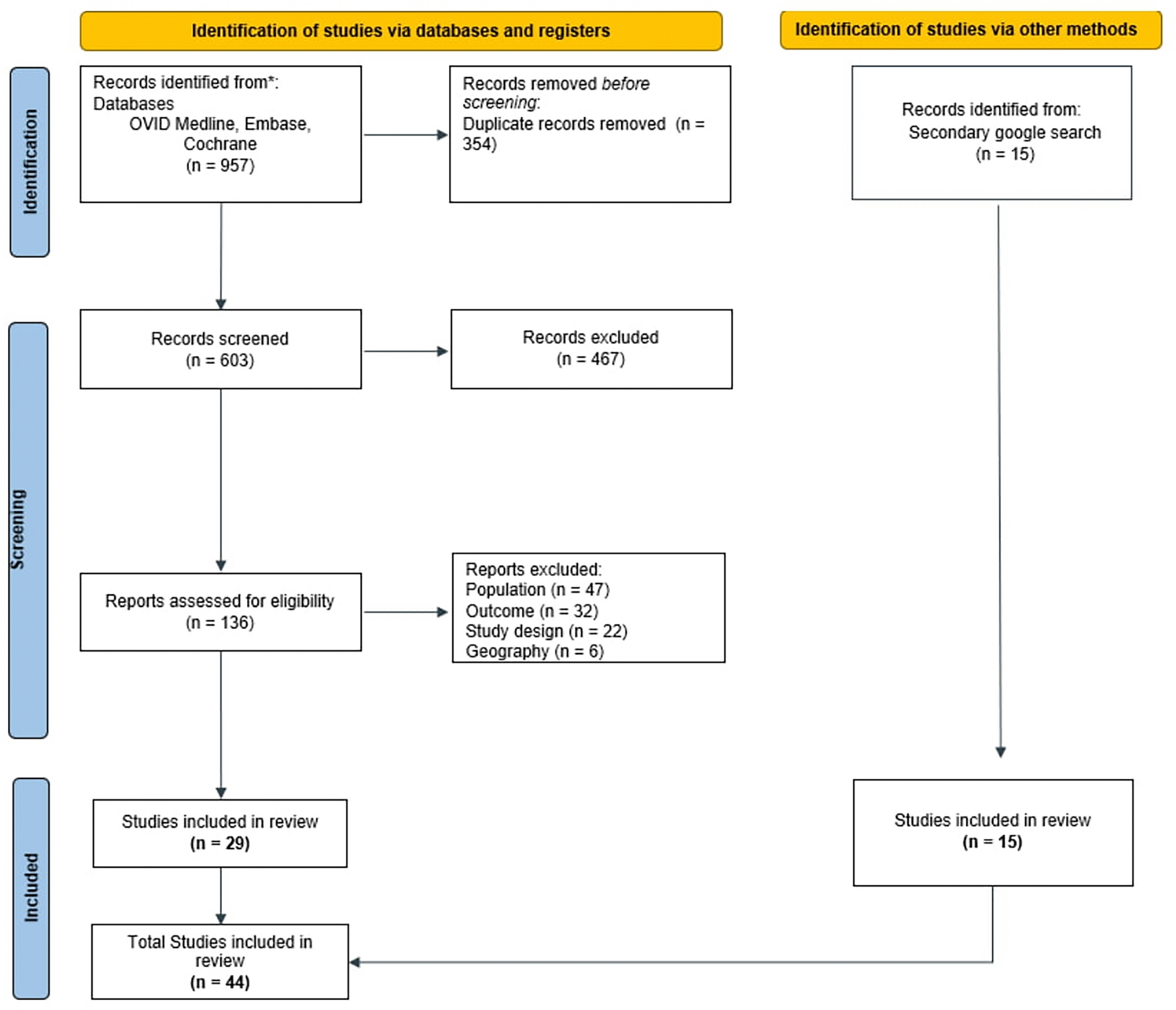
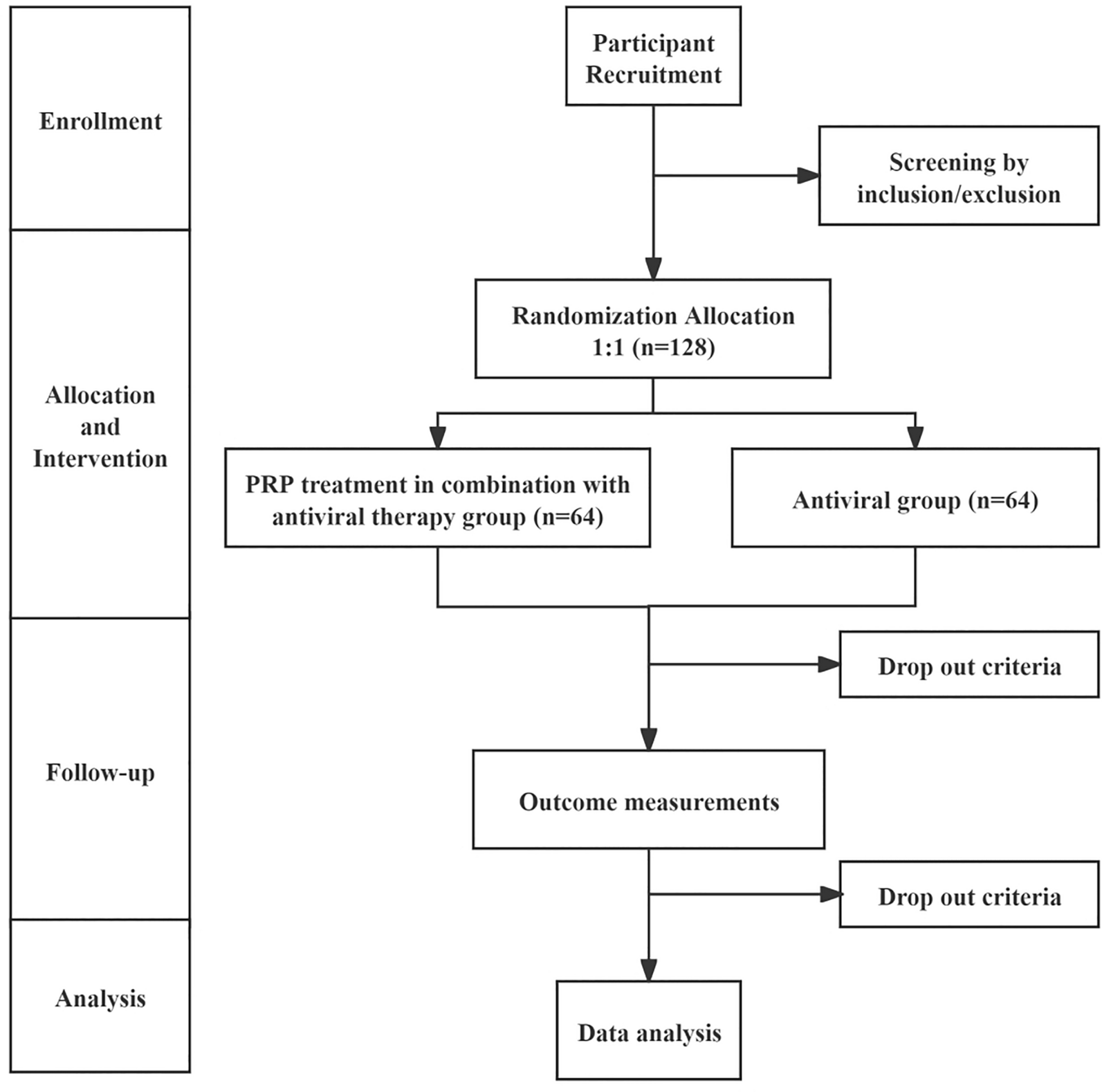
All potential study participants will be provided with a detailed understanding of the study purpose, interventions, potential benefits, possible risks, and countermeasures, and will have at least 1 h to consider whether to join the study. They will be rigorously evaluated according to the inclusion and exclusion criteria. Eligible patients will be enrolled in the study and sign an informed consent form, with the right to withdraw from this study at any time. The confidentiality of participant records will be protected. The patient flow diagram of the clinical trial is illustrated in Fig. 1
Fig. 1
Trial flow diagram. PRP platelet-rich plasma
Inclusion Criteria 1.Diagnosed with HZ involving unilateral thoracic nerves (T1–T12).
2.Age ≥ 50 years and disease duration ≤ 2 weeks.
3.Average pain intensity score (11-point NRS) within 24 h before randomization ≥ 4 points.
4.Agree to participate in this study and be able to sign the informed consent form.
Exclusion Criteria 1.Patients with disseminated HZ or combined with HZ at other locations.
2.Coagulation dysfunction, taking anticoagulant drugs or/and antiplatelet drugs.
3.Systemic infections or infection at the puncture site.
4.Pregnant or lactating women.
5.Patients with immunodeficiency diseases, severe cardiovascular, cerebrovascular, and respiratory diseases, or other serious systemic diseases that prevent cooperation with the treatment.
6.Patients with psychiatric conditions.
7.Patients with thoracic spine deformity or a history of thoracic spine surgery that affects ultrasound positioning.
8.Patients who have contraindications for PRP or are allergic to treatment-related drugs such as famciclovir, gabapentin, pregabalin, nonsteroidal anti-inflammatory drugs (NSAIDs), and tramadol.
9.Patients with a recent history of other pain management treatments
Case Dropout Instructions 1.If a subject develops other comorbidities, complications, or special physiological changes during the trial and is deemed unfit to continue, they will be classified as a case dropout.
2.If a subject encounters a serious or key adverse event during the trial, and our research team judges it inappropriate to continue, the subject will be withdrawn.
3.If a subject adds medications not specified in the protocol or receives external treatment, it will be considered a protocol violation, and they must be removed from the trial.
4.Subjects may choose to withdraw due to personal reasons or be lost to follow-up in the final stage of the trial.
For all cases of dropout, our research team will actively complete the last follow-up and examination to collect data and analyze effectiveness and safety. Detailed information on all dropout cases, including test results and reasons for dropout, will be fully documented on the case report form.
Randomization and BlindingParticipants will be randomly assigned to receive either PRP treatment in combination with antiviral therapy or antiviral therapy alone in a 1:1 ratio using block randomization with a block size of 4, created with IBM SPSS Statistics 25.0. This sequence will be secured in numbered, opaque envelopes to ensure allocation concealment. Treatment efficacy and safety evaluations will be conducted by independent assessors unaware of the group assignments.
Due to the nature of the interventions, complete blinding is not possible for the doctors and patients. Therefore, this study will use blinded endpoints. The efficacy and safety evaluations will be performed by personnel not directly involved in the treatment. The evaluators who perform the assessments and the personnel conducting the statistical analysis will be blinded to the group assignments.
InterventionsPRP PreparationIn this study, we employed a classical two-step centrifugation method to prepare 3 ml of PRP for paravertebral injection therapy. The PRP used in this study is classified as pure PRP (P-PRP), which contains a high concentration of platelets but minimal leukocytes, following the classification by Dohan Ehrenfest et al.[24].
1.Blood collection: Approximately 30 ml of venous blood is drawn from each patient using sodium citrate as an anticoagulant to inhibit coagulation.
2.First centrifugation: The collected blood undergoes a first centrifugation at room temperature at low speed (160–200 g) for 15 min, resulting in a separation into three distinct layers: a lower red blood cell layer, a middle buffy coat rich in white blood cells, and an upper plasma layer with a low platelet count.
3.Platelet enrichment: The upper layer of plasma, excluding the white blood cell-rich buffy coat, is gently transferred to a new sterile centrifuge tube, taking care to avoid disturbing the middle layer.
4.Second centrifugation: This plasma is then centrifuged at a higher speed (500–600 g) for 10 min to further concentrate platelets at the bottom of the tube.
5.PRP collection: After centrifugation, the upper plasma is carefully aspirated, leaving about 4 ml of PRP at the bottom of the tube. From this, 1 ml is used for platelet counting and activity analysis to ensure the prepared PRP meets clinical standards, and the remaining 3 ml is utilized for ultrasound-guided paravertebral injections.
Ultrasound-Guided Thoracic Paravertebral Injection TechniqueWe adopted an ultrasound-guided approach for thoracic paravertebral injection, as detailed in Pu et al. [25]. The patient will be placed in the prone position to optimize access to the thoracic paravertebral space. A low-frequency convex array ultrasound probe (2–5 MHz) will be used to localize the target thoracic levels, starting in the paraspinal sagittal position and moving caudally to identify the ribs. After identifying the first rib, the probe will be moved further down the thoracic spine to locate the target segments. Typically, one to three thoracic levels will be selected based on the dermatomal distribution of ZAP, with emphasis on the most affected segments (e.g., T3, T4, and T5 for T3–T5 involvement).
Once the target level is identified, the probe will be rotated 90 degrees to achieve an axial view, allowing visualization of the transverse process, inferior articular process (IAP), and parietal pleura (PP). A 22-gauge, 80–100-mm echogenic needle will be inserted in-plane from lateral to medial, aiming for the midpoint between the IAP and PP. Doppler ultrasound will be used to avoid puncturing vascular structures. After confirming the needle placement, 3 ml of PRP will be injected at each targeted level under continuous ultrasound guidance.
Following the injection, patients will be monitored for 30 min to observe any adverse effects. A follow-up ultrasound will be performed to verify the distribution of the injectate and exclude complications such as hematomas or pleural punctures.
Treatment Interval and Treatment DosePatients in both groups will receive standard antiviral therapy with famciclovir 500 mg 3 times daily for 7 days, initiated immediately upon recruitment during the viral outbreak [26]. In addition to the standard antiviral therapy, patients in the experimental group will receive PRP injections. The interval between each ultrasound-guided thoracic paravertebral injection is one week, with a total of four injection treatments. Each injection includes 3 ml of PRP per affected thoracic nerve.
Additional InterventionAntiviral therapy has been widely used as the standard treatment for HZ [27]. During the treatment process, pain management will be adjusted based on pain severity and nature of the pain. For mild pain, non-opioid analgesics, such as NSAIDs, will be administered, with opioids added if the pain progresses to moderate or severe levels. Given the neuropathic component of ZAP, tricyclic antidepressants or anticonvulsants, such as gabapentin or pregabalin, may also be incorporated to enhance pain relief [28]. If the ultrasound-guided PRP injection is unsuccessful or the treatment cannot be completed for any other reason, the corresponding patient will be considered a protocol violation and must be excluded from the study.
After completing the study, the doctor will decide whether to continue auxiliary medications or other treatments based on the patient's specific condition. The dosage of medication will be adjusted according to changes in the patient's pain level, aiming to optimize therapeutic effects and improve quality of life. Treatments not mentioned in this protocol will not be permitted unless approved and standardized by the research team. All additional treatments will be documented in detail by the research assistant.
Follow-up and Participant TimelineAfter patients receive therapy, systematic follow-up will be conducted. Follow-up time points are shown in Table 1.
Table 1 Timeline of participant enrolment, allocation, interventions, and outcome assessmentsOutcome AssessmentBaseline Demographic CharacteristicsBaseline demographic characteristics of the patients will be collected through institutional electronic medical records or direct inquiry to compare with post-intervention outcomes.
Primary OutcomeIncidence rate of PHN: The incidence rate of PHN will be calculated based on the percentage of patients with an NRS score ≥ 4 at the follow-up visits scheduled at 3 months, 6 months, and 12 months.
Secondary Outcomes 1.Pain intensity: Assessed using the NRS-11, where scores range from 0 (no pain) to 10 (worst possible pain).
2.Pain characteristics: Including the presence of symptoms such as itching, paresthesia and hyperalgesia.
3.Skin lesions: Including the number of days from the initial onset of rash to healing, the largest skin lesion area.
4.Quality of life score: Assessed using the 12-item Short-Form Health Survey (SF-12) [29].
5.Sleep quality: Evaluated using the Pittsburgh Sleep Quality Index (PSQI) [30].
6.Analgesic consumption: Average weekly consumption and name of rescue analgesics for each patient.
7.Adverse events and safety assessment: Comprehensively record and analyze all treatment-related adverse events, including intra-treatment and post-treatment complications and adverse reactions.
Statistical AnalysisAll analyses will be conducted on an intention-to-treat (ITT) and per-protocol (PP) basis to ensure the robustness of the results.
Primary OutcomeA chi-square test will be used to compare the incidence of PHN between the PRP treatment group and the antiviral therapy group. Additionally, the relative risk (RR) and its 95% confidence interval will be calculated to evaluate the relative risk of PRP treatment compared to antiviral therapy.
Secondary OutcomesFor quantitative data (such as NRS-11, SF-12, and PSQI scores), the t-test or Mann–Whitney U test will be used to compare the differences between the two groups according to the data distribution characteristics. For categorical results (such as complication rate), chi-square test or Fisher's exact test will be used. All statistical analyses will use two-sided tests, with the significance level set to 0.05. For continuous repeated measurement data, such as quality of life scores at different time points after treatment, repeated measures ANOVA or mixed models will be used to analyze data for time effects and between-group effects.
Sample Size CalculationBased on the reported prevalence, the incidence of PHN after antiviral therapy with famciclovir after VZV relapse in patients > 50 years of age is approximately 34.8%[31]. We predict a similar incidence rate in the control group. For the PRP group, our clinical experience suggests an estimated incidence of 10%. To achieve a 90% confidence level and a 5% significance level, we used PASS V.15 software (NCSS, Kaysville, UT, USA) to estimate that a minimum of 58 patients per group is required. Considering a potential 10% dropout rate, we plan to recruit 64 patients per group, totaling 128 patients.
Handling of Outliers and Missing DataTo address missing data, multiple imputation will be utilized. Outliers will be analyzed based on their origin and nature to determine whether correction, removal, or the application of data transformation methods is necessary to mitigate their impact.
Data Collection and ManagementStudy conduct and data collection: This study will be conducted by specially trained research assistants responsible for collecting data from patients who meet the inclusion criteria. We will employ an Electronic Data Capture (EDC) system to collect baseline data prior to treatment, including demographic characteristics, NRS scores, SF-12, and PSQI, among others. To ensure data accuracy and confidentiality, all data will be initially recorded on paper Case Report Forms (CRFs), then verified and entered into the EDC by data management personnel.
DSMC review: The Data Safety Monitoring Committee (DSMC) will conduct regular reviews of the data to ensure the safe progression of the study and promptly identify any potential safety issues.
Data access and protection: All researchers accessing the data are required to sign a confidentiality agreement and undergo training on data protection regulations.
Adverse events (AEs): AEs will be meticulously documented in the CRFs and assessed by researchers for causality with the treatment. The nature, timing, duration, severity, and outcome of AEs will be recorded and evaluated by physicians. All AEs will be promptly addressed and reported to the study monitor and Institutional Review Board (IRB), especially those that are serious or life-threatening. The aim is to address any issues promptly, including halting the study if necessary.
留言 (0)