Ethic statement
The study follows the guidelines of the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85 − 23, revised in 1996). All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of Jinan University and Sun Yat-sen University Cancer Center. Balb/c nude mice used in this study were ordered from the Charles River Company (Beijing, China) and housed under specific-pathogen-free conditions.
Cell culture
HCT116, 786O, HEPG2, A549 and FHC cells were purchased from ATCC. All cell lines were maintained in an incubator at 37℃ with 5% CO2. HEPG2, 786O, A549, FHC, and HCOEPIC cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (GIBCO, USA) supplemented with 10% Fetal Bovine Serum (FBS), 100 U/ml penicillin, and 100 mg/ml streptomycin sulfate. HCT116 cells were maintained in RPMI 1640 medium (GIBCO, USA) supplemented with 10% FBS, 100 U/ml penicillin, and 100 mg/ml streptomycin sulfate. All cultured cells were regularly tested and found to be mycoplasma free.
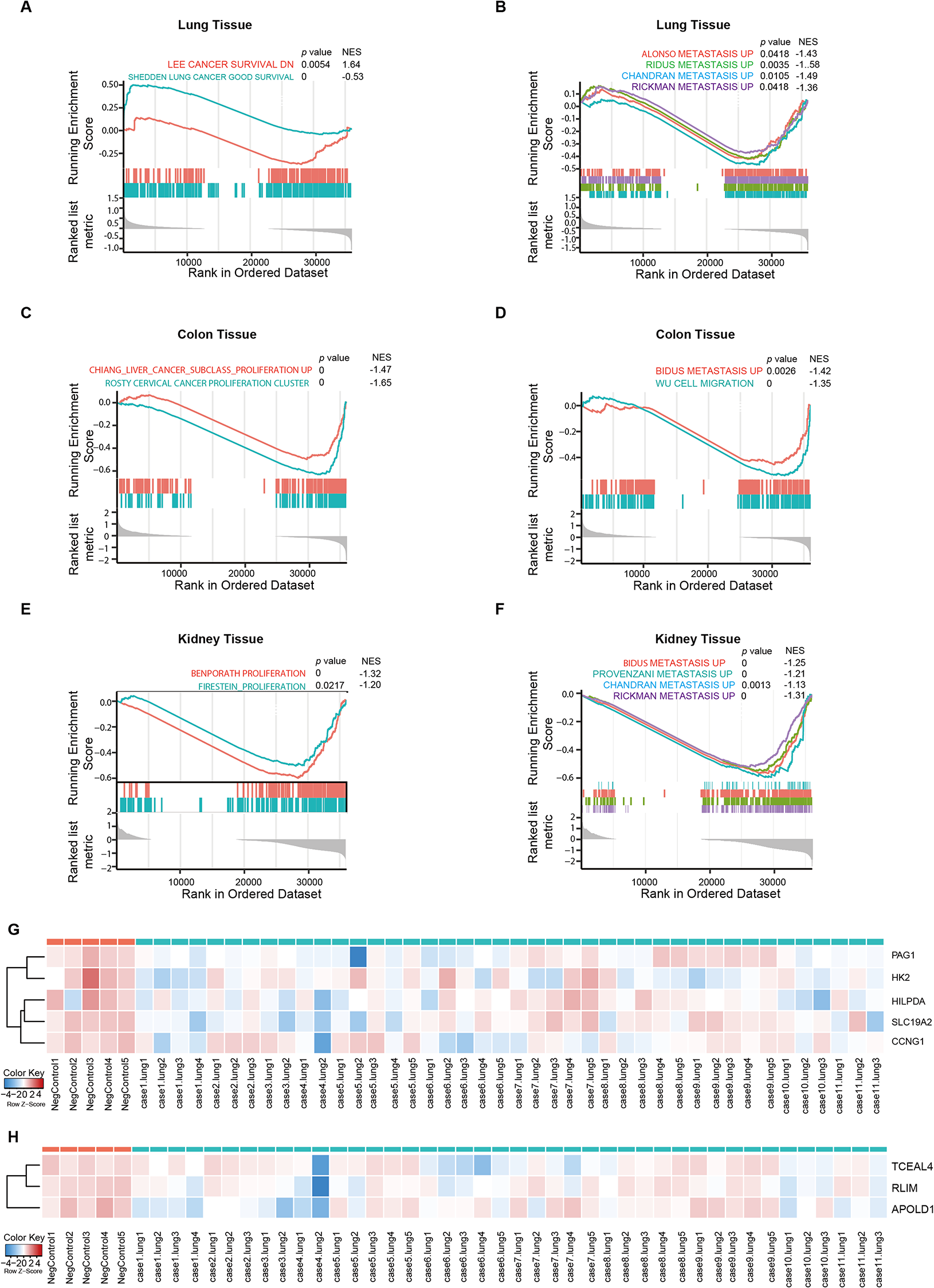
Gene set enrichment analysis (GSEA)
GSEA was applied to evaluate enriched cellular pathways and relevant molecular mechanisms. The GSEA software (version 3.0) was downloaded from GSEA website. A subset of c2.cp.kegg.v7.4.symbols.gmt was downloaded from the Molecular Signatures Database.
Colony-formation assay
Five hundred cells were seeded into each well of 6-well plates. Following a cultivation period of 10–14 days, the colonies were immobilized, subjected to staining, and subsequently enumerated.
Transwell assays
For the transwell experiments, 500 µl of cell culture medium with fetal bovine serum (FBS) was introduced into the lower compartment of the chamber (BD Biosciences, USA). 5 × 104 − 2 × 105 cells suspended in 500 µl serum-free medium were subsequently plated into the upper insert of a 24-well plate. Following an incubation period of 24–36 h, the cells that had invaded were immobilized, subjected to staining, and subsequently enumerated under a microscope. All images were processed using ImageJ software.
Animal experiment
In various experimental models, animals were chosen at random, ensuring a minimum of five animals within each condition. All BALB/c nude mice (female, 18–20 g, four weeks old), utilized in the lung metastasis model, the bone metastasis model, and the subcutaneous xenograft model, were procured from Charles river Laboratory (Beijing, China).
In the subcutaneous xenograft model, a total of 5 × 106 HCT116 cells, HEPG2 cells or 786O cells were introduced into the right flank of each four-week-old BALB/c nude mouse. Tumor formation was assessed after 4–6 weeks.
For the lung metastasis model, 1 × 106 luciferase-transduced HCT116 cells or 4 × 106 A549 cells were injected into the tail vein of each four-week-old BALB/c nude mouse. Four to six weeks post-injection, the mice were sacrificed. The lungs were obtained and after fixation, embedding, and H&E staining, the number of metastatic lesions were calculated to evaluate tumor growth. To visualize the metastatic tumors, the IVIS 200 imaging system was employed after anesthetizing the mice with isoflurane and administering 100 µl of intraperitoneal VivoGlo™ luciferin solution (Promega, USA).
For bone metastasis model, 5 × 106 luciferase-transfected A549 cells transfected with N-Flag or control plasmids were injected into the left cardiac ventricle after anesthetized. 8 weeks later, the metastatic condition of bone was measured by IVIS 200 imaging system. Then the mice were euthanized and the marked bones were acquired and conducted H&E staining.
To determine the in vivo effect of N protein on tumor metastasis, 5 × 105 luciferase-transduced HCT116 cells were injected into the tail vein of 10 four-week-old BALB/c nude mice which were randomly divided into two groups. After 7 days when the inoculated tumor cells formed colonies in the lung, we directly intravenously injected purified SARS-CoV-2 N protein or PBS in the tail vein of BALB/c nude mice. Animals were given single injections with 10 mg/kg twice a day. After one month, the metastatic tumors were measured by IVIS 200 imaging system and then euthanized.
RNA immunoprecipitation (RIP)
The RNA Immunoprecipitation (RIP) procedure was executed following the guidelines of the Magna RIP RNA-Binding Protein Immunoprecipitation Kit (Millipore, USA). Cells were gathered and subjected to lysis buffer for a duration of thirty minutes at 4 °C. After centrifugation, the resulting lysates were collected and allowed to incubate overnight with 30 µl of Protein-A/G beads (Thermo Fisher Scientific, USA) with the appropriate antibodies. The beads were washed for 6 times. Before the immunoprecipitated RNA was extracted using TRIzol reagent (Invitrogen, USA) for subsequent sequencing analysis. The primary antibodies used for the RIP process in our investigation were as follows: anti-Flag-HRP (1:200, Cell Signaling Technology, USA, catalog number: 2368); anti-YBX1 (1:100, Abcam, catalog number: ab76149).
Preparation of RNA libraries and high-throughput sequencing
The quality and quantity of RNA was assessed by Agilent 2200 TapeStation (Agilent Technologies, USA) and Qubit (Thermo Fisher Scientific, USA). Briefly, RIP RNAs were fragmented to approximately 200 bp. Then, the RNA fragments were subjected to cDNA synthesis and adaptor ligation following the instructions of NEBNext® Ultra RNA LibraryPrep Kit for Illumina (NEB, USA). The final library product was assessed with Agilent 2200 TapeStation and Qubit® (Life Technologies, USA) and then sequenced on Illumina (Illumina, USA) platform with pair-end 150 bp at Ribobio Co. Ltd (Ribobio, China). Date preprocessing Adaptor and low-quality bases were trimmed with Trimmomatictools (version:0.36), and the clean reads undergone rRNA deleting to get effective reads. Genomic alignment (version from UCSC genome browser) was using Tophatb (version:2.0.13) to get unique mapping reads.
Peaks calling and motif identification in RIP-Seq
Piranha (version 1.2.1) was employed to perform peak calling. Then using Hommer (version:4.8) to annotate the Peaks. The nucleotides in Peaks region were used for detection of the consensus motif by STREME (version: 5.3.0) and MEME (version:5.3.0). Motif central enrichment was performed by CentriMo (version:5.3.0).
Functional enrichment analysis
Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis were performed using KOBAS3.0/ the clusterProfiler package. The enriched results were restricted to GO biological process and KEGG pathway terms. The GO biological process and KEGG pathway terms with adjusted P-value < 0.05 were considered to be significant.
Coimmunoprecipitation and mass spectrometry
Protein extraction from plasmid-transfected cells was carried out, and 1/10 protein lysis solution of each sample was obtained as input. The rest protein lysis solution was incubated with Flag (1:200, Cell Signaling Technology, USA, catalog number: 2368) antibodies followed by a 30-minute rotation at 4 ℃. Subsequently, samples were subjected to incubation with Sepharose-conjugated protein G magnetic beads (Thermo Fisher Scientific, USA) for overnight at 4 °C. After 4 times washing, the beads were suspended in 1× SDS and boiled for 10 min. Western blotting was then conducted to verify the IP efficiency. Mass spectrometry procedures were conducted by Wininnovate Bio (Shenzhen, China). Tandem mass spectrometry data were obtained using data-dependent acquisition mass spectrum techniques via a ThermoFisher Q Exactive mass spectrometer (Thermo Fisher Scientific, USA).
Western blotting
After PBS wash, the harvest cells were lysed by RIPA buffer. After 12,000 rpm centrifuge for 15 min, the supernatants were collected. The supernatant protein lysates subsequently were dissolved in 1× SDS buffer and separated through SDS-PAGE. Upon transferring to a PVDF membrane (Millipore, USA), the membrane was subjected to an overnight incubation at 4 °C with primary antibodies, followed by an additional 1 h incubation at room temperature with secondary antibodies. After washing for 3 times, the signals presenting on the membranes were visualized using an enhanced chemiluminescence kit (Tanon, China). For Western blotting in our study, the primary antibodies used included: rabbit polyclonal anti-Flag-HRP (1:1000, Cell Signaling Technology, USA, catalog number: 2368), PKM1/2 (1:500, Cell Signaling Technology, USA, catalog number: 3106), LDHA (1:1000, Cell Signaling Technology, USA, catalog number: 3582), BCL-2 (1:1000, Cell Signaling Technology, USA, catalog number: 4223), BAX (1:2000, Cell Signaling Technology, USA, catalog number: 5023), Caspase 3 (1:1000, Cell Signaling Technology, USA, catalog number: 9962), Cleaved Caspase 3 (1:1000, Cell Signaling Technology, USA, catalog number: 9661), PARP (1:2000, Cell Signaling Technology, USA, catalog number: 9532), Cleaved PARP (1:2000, Cell Signaling Technology, USA, catalog number: 5625), GAPDH (1:2000, Cell Signaling Technology, USA, catalog number: 2118), ACTIN (1:2000, Cell Signaling Technology, USA, catalog number: 4970).
Protein-protein interaction network
The protein-protein interaction network was built and visualized by using STRING software. The green, red, blue, black, cyan, and purple edges represent the predicted gene neighborhood interactions, gene fusions, gene co-occurrence interactions, co-expression interactions, known interactions from curated databases, and known interactions experimentally determined, respectively. Besides, the different color of nodes represents the included GO terms. The red nodes represent the mRNA processing terms (GO:0006397), the blue nodes represent the mRNA binding terms (GO:0003729).
Quantitative real-time polymerase chain reaction
Total RNA from the samples was extracted using TRIzol reagent (Invitrogen, USA). Subsequently, 1 µg of the extracted RNA underwent reverse transcription to cDNA, following the protocol outlined in the PrimeScript RT reagent kit (TaKaRa, JP). For quantitative real-time polymerase chain reaction (qRT-PCR), SYBR Green SuperMix (Roche, USA) was employed in combination with the ABI Prism 7000 Sequence Detection System (Applied Biosystems). The qRT-PCR data were analyzed using the Relative Quantification (ΔΔCT) method. The primer sequences utilized are listed as bellow:
PKM forward:
5’-AGTACCATGCGGAGACCATC-3’
PKM reverse:
5’-GCGTTATCCAGCGTGATTTT-3’
GAPDH forward:
5’-CAGCCTCAAGATCATCAGCA-3’
GAPDH reverse:
5’-ATGATGTTCTGGAGAGCCCC-3’
Immunofluorescence
Cultured cells were washed three times with cold PBS, and fixed with 4% paraformaldehyde (PFA) for 15 min. After 10 min incubation with 0.5% Triton X-100, cells were blocked with 5% BSA for 1 h at room temperature. They were then subjected to an overnight incubation with the primary antibody at 4 °C. On the next day, the cells were exposed to the corresponding secondary antibody for 1 h at room temperature and subsequently stained with DAPI. The images were captured using a Leica confocal microscopy system. The antibodies used in immunofluorescence were as follows: anti-Flag (Cell Signaling Technology, USA, catalog number: 2368) at a dilution of 1:100, and anti-G3BP1 (Abcam, UK, catalog number: ab181150) at a dilution of 1:200.
mRNA stability
10 µg/ml actinomycin D (ActD) was directly added into cells for the indicated times. The cells were harvested at indicated time after addition of ActD. mRNA amounts were measured by qRT-PCR as described above and normalized to Actin before calculation of half-lives.
Apoptosis analysis
Cultured cells were washed three times with cold PBS. Assessment of apoptosis was carried out using an Annexin V Apoptosis Detection Kit (Thermo Fisher Scientific, USA), following the manufacturer’s instructions. In brief, the cells were suspended in 100 µL of binding buffer containing 5 µL of FITC-conjugated Annexin V antibody, followed by a 15-minute incubation at room temperature. Subsequently, they were washed and resuspended in 200 µL of binding buffer, to which 5 µL of propidium iodide (PI) was added. Finally, the cells were subjected to analysis using the CytoFlex flow cytometry system (Beckman Coulter, USA) and the FlowJo software (Treestar).
Seahorse experiment
XF96 analyzer (Agilent, Santa Clara, CA, USA) was used to conducted XF glycolysis stress test and mitochondrial stress test according to the manufacturer’s instructions. Briefly, cells were seeded in Seahorse XF plates at a density of 5 × 104 per well and cultured for 24 h. The low buffered XF assay medium (Agilent, JP), supplemented with 10 mM glucose and 2 mM glutamine was used in the next day. Cells were then cultured for 1 h at 37 °C in a no-CO2 incubator. Seahorse XF analysis, Oxygen Consumption Rate (OCR = pmole O2/min) and ExtraCellular Acidification Rate (ECAR = mpH/min) were conducted at 37 °C.
Plasmid construction and transfection
The N gene of SARS-CoV-2 (GenBank accession number MN908947.3) were synthesized by GenScript, Nanjing, China, and cloned to pcDNA3.1(+) or plv lentivirus-expression vector with Flag-tag. The truncated DNA fragment of N mutant were amplified from pcDNA3.1(+)-N-Flag plasmid and inserted into pcDNA3.1(+) or plv lentivirus-expression vector with Flag-tag. Plasmid transfection was conducted using Lipofectamine 2000 (Invitrogen, CA, USA) or JetPEI (Polyplus).
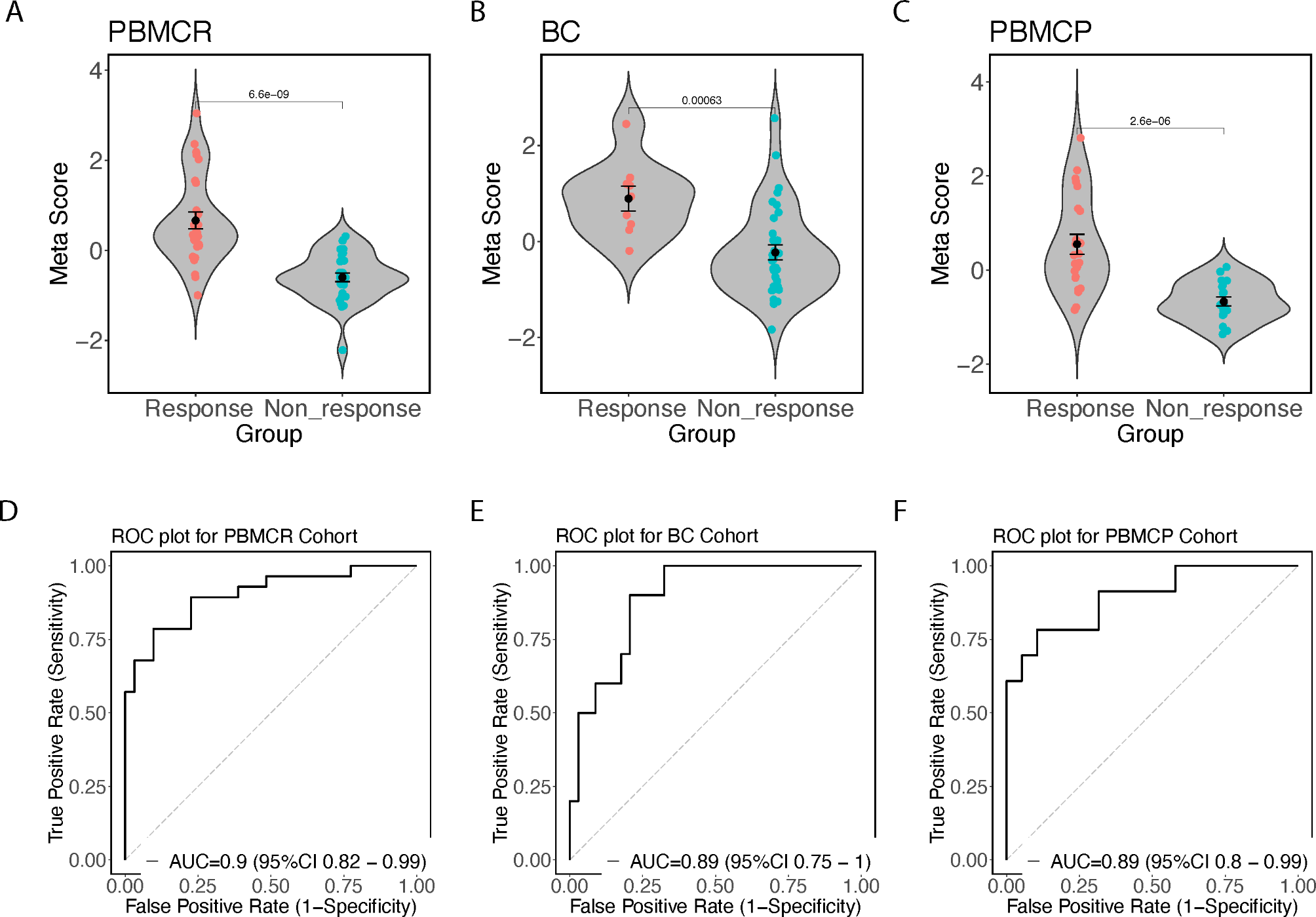
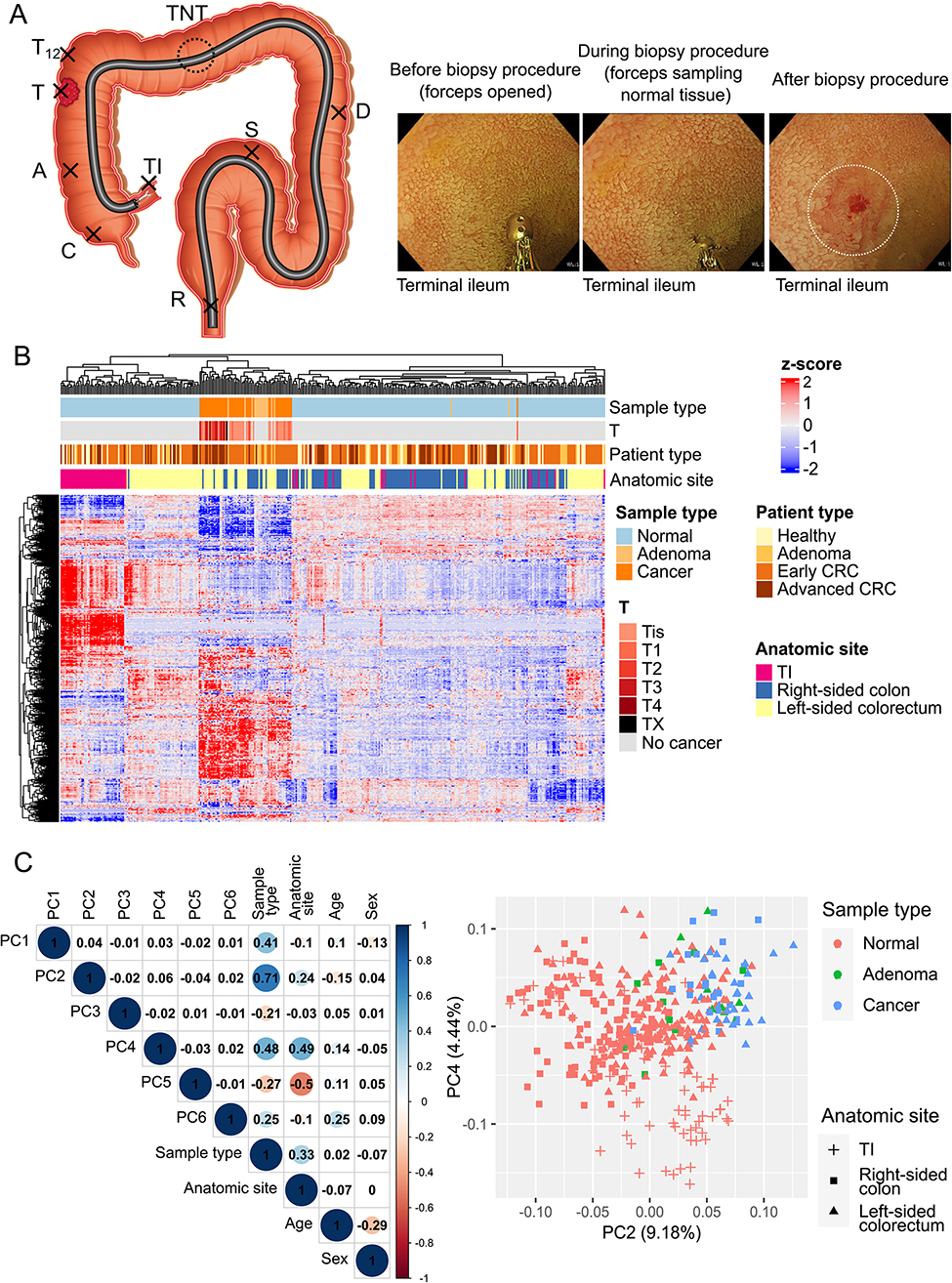
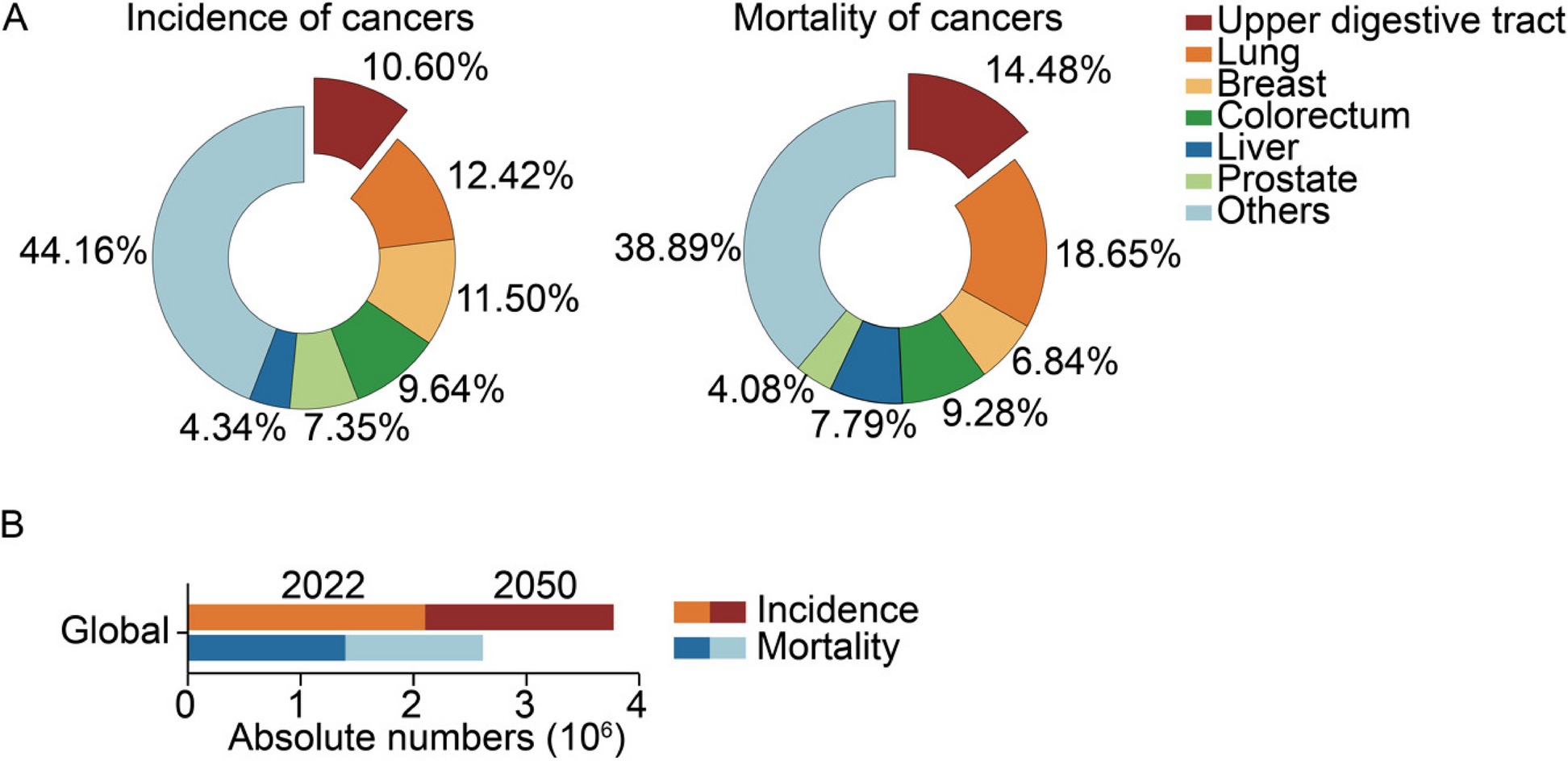
Quantification and statistical analysis
All statistical analyses are described in the corresponding methods sections and indicated in the figure legends. Statistical analysis was carried out using GraphPad Prism 10. All results are presented as the mean ± SD or SEM. NS, no significance.




留言 (0)