
記住我

The human cancer cell lines HCC827 (KRASWT), NCI-H292 (KRASWT), A549 (KRASG12S), NCI-H1299 (NRASQ61K), NCI-H460 (KRASQ61H), PANC-1 (KRASG12D), HCT116 (KRASG13D), SW480 (KRASG12V) and the human embryonic kidney cell line HEK293T were obtained from American Type Culture Collection (ATCC). The human cancer cell line PA-TU-8902 (KRASG12V) was obtained from Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ). The human lung epithelial cell line BEAS-2B and the human pancreatic duct epithelial-like cell line hTERT-HPNE were obtained from FuHeng Biology. The human cancer cell line NCI-H2030 (KRASG12C) was obtained from Zhong Qiao Xin Zhou Biotechnology. The human cancer cell line PC9 (KRASWT) was a kind gift from Dr. Q. Dong (China State Key Laboratory of Oncogenes and Related Genes). All the cell lines were authenticated routinely via short tandem repeat (STR) DNA profiling and tested for mycoplasma contamination every 6 months using MycoBlue Mycoplasma Detector (Vazyme).
BEAS-2B, HEK293T, HPNE, PANC-1 and PA-TU-8902 cells were propagated in DMEM medium (BasalMedia), A549 cells were propagated in F-12K medium (BasalMedia), HCT116 cells were propagated in McCoy’s 5a medium (BasalMedia), SW480 cells were propagated in Leibovitz’s L-15 medium (BasalMedia), and HCC827, PC9, NCI-H292, NCI-H1299, NCI-H460 and NCI-H2030 cells were propagated in RPMI-1640 medium (Gibco). All media were supplemented with 10% fetal bovine serum (Biological Industries), 1% penicillin‒streptomycin (BasalMedia) and 1% GlutaMax (BasalMedia).
Construction of DOX-inducible shNPPS cell linesA lentivirus system was used to construct doxycycline-induced shNPPS cell lines. The inducible shRNA plasmids targeting NPPS (GV307 vector) were obtained from GeneChem, and their sequences were based on siNPPS #4 and #5. In brief, lentiviruses were generated in HEK293T cells by cotransfecting the shNPPS plasmid and virus-packaging vectors (pSPAX2 and pMD2.G) and then the viral supernatants were harvested after 24 h and 48 h to infect the target cells. Positive cells were selected with puromycin (1 μg/mL) after 48 h of infection.
Isogenic H292 cellsUsing homologous recombination (HR)-mediated gene knock-in based on CRISPR/Cas9 technology, we inserted the KRAS p.G12C gene variant into the H292 cell line. In brief, the KRAS knock-out plasmid (pX459-sgKRAS) and the KRAS G12C donor plasmid (pUC19-KRASG12C) were kindly provided by Dr. Y. Shen (Shanghai Jiao Tong University School of Medicine). H292 cells were seeded in 6-well plates. The next day, the cells were cotransfected with the sgKRAS plasmid and the donor plasmid using Lipofectamine 3000 (Invitrogen). After 48 h, cell media were supplemented with puromycin (1 μg/mL), and single-cell colonies were subsequently established via dilution in 96-well plates. After 10 days, single cell clones were transferred to 24-well plates and expanded. For sequence analysis, genomic DNA was extracted from single cell clones using the Genomic DNA Mini-Preps Kit (Sangon Biotech) and PCR products were amplified using the forward primer 5ʹ-AGCGTCGATGGAGGAGTTTG-3ʹ and the reverse primer 5ʹ-GACCCTGACATACTCCCAAGG-3ʹ. The PCR products were subsequently sequenced using the forward primer (BioSune Biotechnology) and sequence analysis was conducted using SnapGene software.
siRNA and plasmid transfectionFor siRNA transfection, cells were plated at a confluence of 30%–40% when they were transfected with a specific-targeted siRNA duplex using Lipofectamine 3000 (Invitrogen) according to the manufacturer’s instructions. As control, cells were transfected with a noncoding siRNA (NC). The final concentration of the siRNAs was 20 nM. The siRNAs were obtained from Shanghai GenePharma. The NPPS siRNA sequences from 5ʹ to 3ʹ were as follows:
siNPPS #4: -AGATAAATACTATTCATTT-
siNPPS #5: -AGCTTCTATCAACAAAGAA-
For plasmid transfection, cells were plated in 6 cm dishes at a confluence of 60%–80% when they were transfected with 500 ng of plasmid using Lipofectamine 3000 (Invitrogen) according to the manufacturer’s instructions. The cells were transfected with the empty vector as a negative control. Plasmids for human NPPS (NM_006208.3), human KRAS (NM_004985), KRAS G12C (G12C mutation), KRAS G12D (G12D mutation), and KRAS Q61H (Q61H mutation) with a C-terminal 3 × Flag (CV702 vector), and plasmids for human HK1 (NM_000188.3), HK1-∆MBD (1–21 aa deletion), HK1-∆N’ (22–474 aa deletion), and HK1-∆C (475–917 aa deletion) with a C-terminal HA (GV366 vector) were constructed by Shanghai GeneChem.
Cell growth and cell viability assaysA total of 3000–4000 cells were seeded in 96-well plates. The next day, the cells were transfected with siRNAs or treated with drugs at the indicated dosages. Cell growth was monitored by IncuCyte ZOOM live cell analysis system (Essen BioScience) every 4 h for 72‒96 h. Cell viability was determined via Cell Counting Kit-8 (CCK-8; Proteintech Group) according to the manufacturer’s instructions.
For some experiments, the cells were fixed with 4% paraformaldehyde and stained with 0.1% crystal violet after the indicated treatment. The plates were scanned by Epson Scanner (Seiko Epson Corporation). The crystal violet in the plates was dissolved in 10% acetic acid, and the optical density at a wavelength of 600 nm (OD600) was detected on a microplate reader (Thermo Fisher Scientific).
Inhibitors and antibodiesThe following compounds were used in this study: 2-Deoxy-D-glucose (Selleck, S4701), Enpp-1-IN-1 (Selleck, S0501), Sotorasib (Aladdin, S414206), and Doxycycline Hyclate (Selleck, S4163).
The following antibodies were used in this study: NPPS (Abcam, ab40003), NPPS (Abcam, ab223268), HK1 (Cell Signaling Technology, #2024), pERK1/2 (Cell Signaling Technology, #4370), ERK1/2 (Cell Signaling Technology, #4695), pAKT (S473) (Cell Signaling Technology, #4060), AKT (Cell Signaling Technology, #4691), HA (Cell Signaling Technology, #3724), β-actin (Proteintech Group, 66009-1-Ig), and Flag (Sigma‒Aldrich, F1804).
Western blot analysisThe cells or tumor samples were lysed on ice with RIPA lysis buffer (Beyotime) supplemented with protease inhibitor (Beyotime). The protein concentration was measured by BCA Protein Assay Kit (Thermo Fisher Scientific) and adjusted to approximately 2 μg/μL. Samples containing 40 μg of protein were separated by SDS‒PAGE and then transferred to PVDF membranes (Millipore) followed by blockage with 5% nonfat milk for 1.5 h at room temperature. The membranes were then incubated with primary antibodies at 4 °C overnight. The next day, the membranes were incubated with HRP-conjugated secondary antibodies (Beyotime) for 1.5 h at room temperature. Immunoblots were developed via enhanced chemiluminescence (ECL) (Thermo Fisher Scientific) and scanned with the Odyssey Fc imaging system (LI-COR Biosciences).
Coimmunoprecipitation (Co-IP)The cells were lysed on ice with NP-40 lysis buffer (Beyotime) supplemented with protease inhibitor (Beyotime). The protein concentration was measured by BCA Protein Assay Kit (Thermo Fisher Scientific) and samples containing 1–1.5 mg of protein were used for immunoprecipitation.
For IP of endogenous proteins, lysates were incubated with anti-NPPS or anti-HK1 at 4 °C with rotation overnight. The next day, Protein A/G Magnetic Beads (Millipore) were added to the lysates to capture the immunocomplex for 3 h.
For IP of exogenous Flag-NPPS, lysates were incubated with anti-FLAG M2 Magnetic Beads (Sigma–Aldrich) at 4 °C with rotation overnight.
After incubation, the beads were washed for 5 times and resuspended in 2 × SDS loading buffer (Beyotime) followed by boiling at 100 °C for 5 min. Co-IP proteins were evaluated via Western blot analysis.
Protein mass spectrometryThe immunoprecipitates pulled down with anti-NPPS from H292 samples were used for protein mass spectrometry. All MS experiments were performed on a QE-Plus mass spectrometer connected to an Easy-nLC2000 via an Easy Spray (Thermo Fisher Scientific). The ion spectra were analyzed using PEAKS 8.0 (Bioinformatics Solutions) for processing, de novo sequencing and database searching. The resulting sequences were searched through the UniProt Human Proteome database (downloaded on May 5th, 2018). For all the experiments, these settings yielded an FDR of < 1% at the peptide-spectrum match level.
RNA-seq analysisFor RNA-seq, RNA was isolated using TaKaRa MiniBEST Universal RNA Extraction Kit (TaKaRa) and then reverse transcribed into cDNA by PrimeScript™ RT reagent Kit (TaKaRa). The samples were library prepped and sequenced via the Illumina HiSeq 4000 platform by BGI Genomics. Differentially expressed gene (DEG) analysis and gene set enrichment analysis (GSEA) were performed using an online-based software Dr. TOM (BGI).
LC‒MS/MS analysisFor untargeted metabolomics, cells were cultured in 6-well plates and collected in 1.5 mL centrifuge tubes. Metabolites were then extracted with 80% ice-cold methanol followed by vortexing for 5 min. After centrifugation at 20,000 × g and 4 °C for 15 min, the supernatant was transferred and evaporated to dryness using a vacuum centrifuge. The samples were resuspended in 200 µL of 50% (v/v) acetonitrile prior to LC‒MS/MS analysis.
LC‒MS/MS was performed using the ExionLC AD UPLC system (SCIEX) coupled with a TripleTOF 6600 Plus mass spectrometer (SCIEX). Data were acquired using Analyst 1.8.4 software (SCIEX). Metabolite identification and quantitation were performed using the SCIEX OS 1.7 software (SCIEX).
SIRM analysisFor glucose metabolism SIRM analysis, the cells were cultured in 6-well plates and the cell medium was changed to glucose-free RPMI-1640 medium supplemented with 10% FBS and 11 mM U-13C6-glucose (Cambridge Isotope Laboratories) 6 h before sample collection. The sample pretreatment process was referred to the LC‒MS/MS analysis. The samples were analyzed via a TripleTOF 6600 plus mass spectrometer (SCIEX) and the data were acquired using SCIEX OS 1.7 software (SCIEX). The incorporation of 13C from U-13C6-glucose induced an intensity shift from the unlabeled position (m + 0) to the labeled position (m + n, where ‘n’ represents the number of incorporated 13C atoms). The natural isotope abundance was corrected by the AccuCor R package (https://github.com/XiaoyangSu/IsotopeNatural-Abundance-Correction).
Seahorse assayThe cells were attached to culture plates at a density of 20,000 cells per well. The extracellular acidification rate (ECAR) was measured with a Seahorse XFe96 Analyzer (Agilent Technologies) according to the manufacturer’s instructions, in the presence of the following compounds: 10 mM glucose, 1 μM oligomycin and 50 mM 2-DG.
HK activity assayHK activity assays were performed using the Hexokinase Assay Kit (Enzyme-linked Biotechnology) according to the manufacturer’s instructions.
Combination effect analysisThe effects were evaluated using a combination index (CI) calculation in accordance with the Chou-Talalay method. The data were analyzed using the CompuSyn software (CompuSyn Inc.): CI = 1, additive effect. CI > 1, antagonistic effect. CI < 1, synergistic effect.
Xenograft assayAll the animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of Shanghai Jiao Tong University School of Medicine and were performed according to the guidelines. For the xenograft assay, approximately 3 × 106 tumor cells were implanted subcutaneously into the flanks of BALB/c nu/nu athymic mice (female, 5 weeks old). Water containing 2 mg/mL doxycycline (DOX) (in 25 mg/mL sucrose, Sangon Biotech) or the control (25 mg/mL sucrose, Sangon Biotech) was administered to each cohort at one day before tumor inoculation. Tumor volumes were measured with a caliper every 2 or 3 days and calculated as 0.5 × length × width2.
GEO database analysisThe gene expression profiles shown in Fig. 1c were obtained from the Gene Expression Omnibus (GEO) database (www.ncbi.nlm.nih.gov/geo; GSE26850).
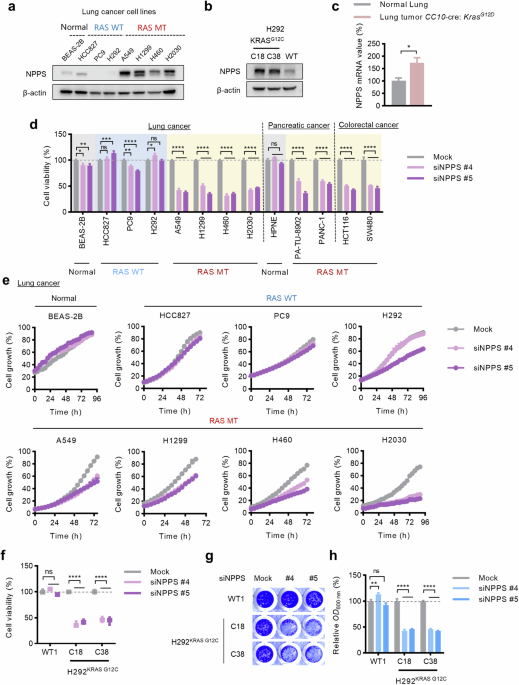
Fig. 1: NPPS is selectively crucial for RAS-mutant cells versus RAS-wildtype cells.
a Protein levels of NPPS in the normal cell line BEAS-2B and cancer cell lines examined by Western blot analysis. b Protein levels of NPPS in H292KRAS G12C cells and H292KRAS WT cells examined by Western blot analysis. c mRNA levels of NPPS in tumors from CC10-Cre/LSL-KrasG12D mice and control murine lung tissues as analyzed via the GEO database analysis (GSE26850). d Cell viability of a panel of cell lines after knockdown of NPPS. The cells were transfected with NPPS siRNAs (20 nM) or mock control for 72 h and then cell viability was determined by CCK-8. e Growth curves of cell lines after knockdown of NPPS. The cells were transfected with NPPS siRNAs (20 nM) or mock control for 72–96 h and monitored by IncuCyte ZOOM system every 4 h. f Cell viability of H292KRAS WT cells and H292KRAS G12C cells after knockdown of NPPS. The cells were transfected with NPPS siRNAs (20 nM) or mock control for 72 h and then cell viability was determined by CCK-8. g, h Cell growth analysis of H292KRAS WT cells and H292KRAS G12C cells after knockdown of NPPS. The cells were transfected with NPPS siRNAs (20 nM) or mock control for 72 h and then cell growth was measured by the crystal violet assay. Data are shown as the means ± SEMs (n = 3 replicates). ns, not significant; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Statistical analysisAll the data are presented as the means ± SEMs and repeated at least 3 times. Statistical analyses were performed using GraphPad Prism software (version 9.0). Significant differences were conducted by 2-tailed unpaired Student’s t test or 1-way or 2-way ANOVA analyses. The statistical parameters can be found in the figures and their corresponding legends. Statistical significance was set as P < 0.05.
留言 (0)