
記住我




As the incidence of diabetes has become globally epidemic, the number of individuals with diabetes has proportionally increased to 425 million, and it is widely predicted to grow until 2045 [1]. Most of the diabetic patients are adults between the ages of 40 and 65 from low and middle-income countries that confer socio-economic status on the affected individual [2]. Diabetes counterfeits desperate organs, expediting macrovascular (coronary artery disease, stroke, etc.) and microvascular complications such as retinopathy, nephropathy, and neuropathy [3]. Approximately 30˗40% of diabetes patients tend to develop nephropathy, causing renal injury and progression to end-stage renal disease [4]. The crucial factors influencing DN include reactive oxygen species (ROS), prolonged hyperglycemia, elevated levels of cytokines, advanced glycation end products (AGEs), enzymatic alteration of protein kinase b (PKB), adenosine monophosphate activated kinase (AMPK), and other metabolic indicators involved in cellular energy metabolism. Additionally, transcription factors also play a critical role in modulating the signaling mechanism involved in hyperglycemia-mediated oxidative stress, leading to the progression of DN [5]. It is well described that DN triggers activation through the functioning of various transcription factors and atypical expression of pro-inflammatory and pro-fibrotic genes in the renal tubulointerstitium [6, 7].
Metabolic memory is defined as an innate mechanism where complications progress to exacerbate despite virtuous glycemic control and glucose normalization [8]. The research history of metabolic memory has been driven by conducting clinical trials and animal experiments. A clinical trial like the Diabetes Control and Complications Trial (DCCT) was conducted in 1983 to obtain the hypothesis of glucose, which led to the conclusion that hyperglycemia is a crucial cause of long-term complications of diabetes [9]. In addition to this, an observational prospective study initiated by the Epidemiology of Diabetes Interventions and Complications (EDIC) predicted the risk association of diabetic complications to be greater in the treatment group compared to the early intensive group [10]. The follow up studies revealed that the levels of HbA1c at the earlier stage can have an impact on long-term complications with no change in the pathological alterations that occurred due to hyperglycemia and persist even after good glycemic control has been achieved [11]. Therefore, the term “metabolic memory” is framed by this conceptual phenomenon of DCCT/EDIC. To date, the theory of metabolic memory and its significance have been enumerated, specific to hyperglycemia, hypoxia, hyperlipidemia, etc. [12]. Finally, the DCCT/EDIC investigations associated with the findings of metabolic memory emphasize the noteworthy and protracted ease of strict glycemic control at the initial stage, driving a substantial change in diabetes management [13]. As the underlying mechanisms of metabolism are still unknown, this literature survey highlights the concept of metabolic memory, its role and action, characteristics, and regulation in hyperglycemia-mediated oxidative stress and renal injury.
Aspects of metabolic memoryMetabolic memory remains persistent and progressive, exhibiting long-term harmful effects such as inflammatory changes, premature cell senescence, apoptosis, etc. A plethora of studies conducted on vascular smooth muscle cells (VSMCs) derived from a diabetes mouse model depicted increased oxidative stress elevating the expression of pro-inflammatory genes and their products, which in turn enhanced the activation of the inflammatory signaling pathway [14]. This phenomenon of persistent metabolic memory negotiates the phenotype of pro-inflammatory VSMCs, causing vascular dysfunction in diabetic individuals. Experimental studies on cultured human endothelial cells exposed to highly concentrated glucose medium showed long-term progression by stimulating the expression of the fibronectin gene even after dispensing the endothelial cells with normal glucose medium. These studies depict the correlation between metabolic memory and divergent expression of antioxidant and inflammatory genes that remain persistent [15]. A rise in AGEs and ROS causes mitochondrial dysfunction after the imbalanced metabolic circumstances in various cells, even after glucose normalization, elevate senescence [16]. The premature senescent cells manifest enhanced metabolic activity, which elevates the deliberation of cytokines, chemokines, and other growth factors, collectively known as the senescence-associated secretory phenotype (SASP). The release of SASP factors causes inflammatory damage and generates detrimentally unambiguous response loop in diabetes [17]. In regard to apoptosis, a sequence of apoptotic genes remains elevated in their expression despite glucose normalization; this condition is assisted by the activation of the pro-apoptotic pathway via metabolic memory [18]. Moreover, there exist three attributions of metabolic memory, the primary one relating to the persistent harmful effect depicting the pro-inflammatory phenotype. Secondary is the early metabolic control showing the long-term impact of metabolic complications; whereas, the latter control is doubtful to have progressive complication prevention. Tertiary impact is the inheritance of long-term adverse effects with progressing metabolic complications to the descending generation; the mechanism behind this defines the development of metabolic memory as closely associated with epigenetic modifications [19].
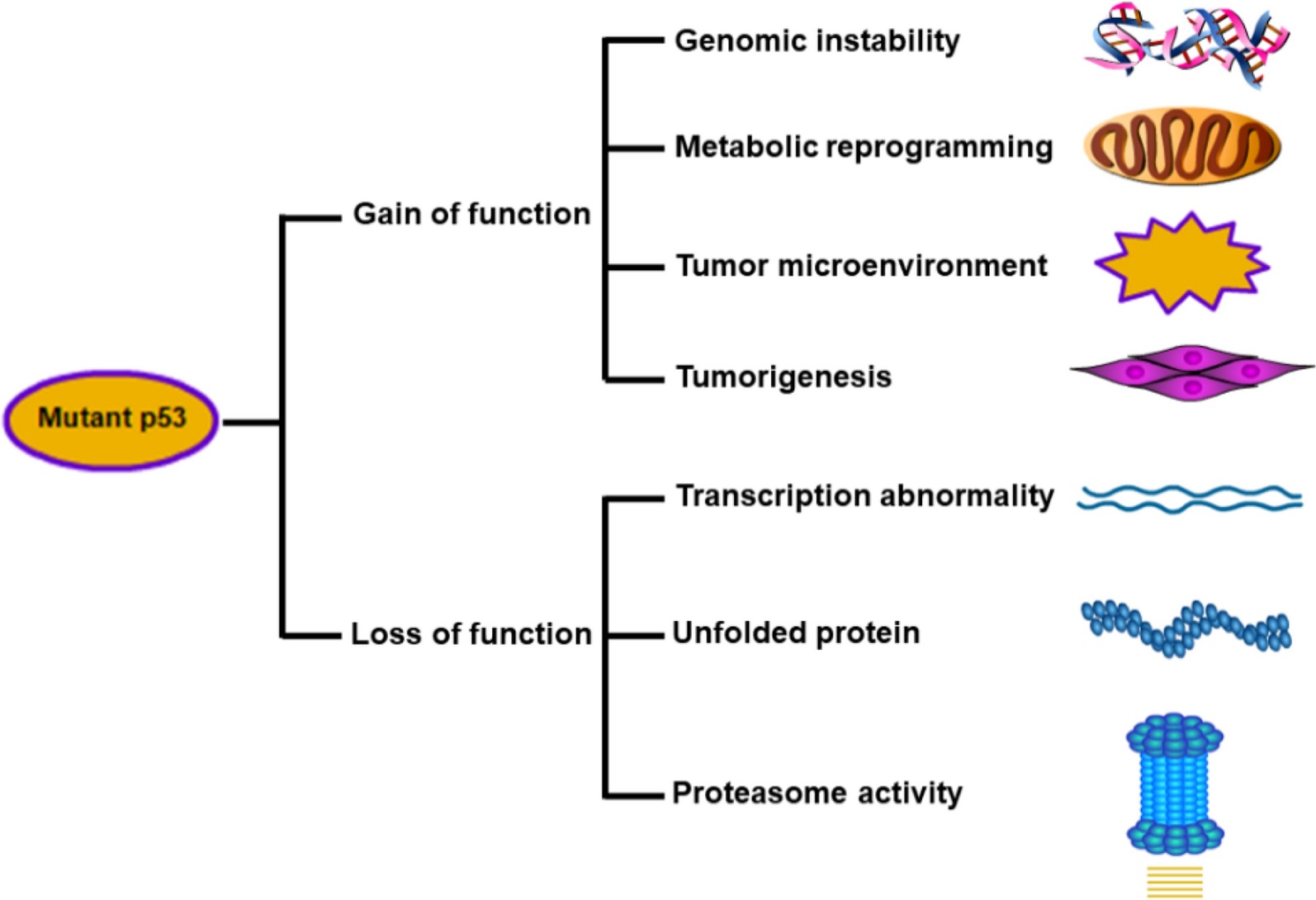
Epigenetic modification as underlying mechanism of metabolic memoryEpigenetic mechanism depends on the modulation of DNA methylation, histone modification, and chromatin remodeling which are interlinked with metabolic memory. Kowluru et al. conducted a set of studies illustrating that short-term existence of hyperglycemia can relatively result in epigenetic changes like DNA methylation, histone modification (methylation, acetylation and deacetylation) represented in Fig. 1 [20, 21]. These changes have depicted to cause perpetual activation of oxidative stress and pro-inflammatory signaling pathways. DNA methylation is well established predominant epigenetic signature to investigate its association with metabolic memory [22]. It is reported that incessant DNA methylation caused due to diabetes induction in zebrafish model, can be disseminated over the cell division process. The fundamental genes related to diabetic kidney injury (renal proximal tubular cells) were also reported to be closely associated with hyperglycemia-mediated metabolic memory by attributing to DNA methylation changes [19, 23]. Yan-Lin wu et al. [24] reported that ten-eleven translocation enzyme-2 (TET2) is thought to have obtained the transforming growth factor beta 1 (TGF-β1) synthesis by reducing its methylation levels, hence promoting the onset of DN. When combined, the up-or down-regulation of DNA methylation levels takes place in an array of tissues and organs, displaying diverse effects and leading to diabetes. Furthermore, obtaining blood and monitoring the levels of DNA methylation are feasible. Even more, site-specific histone modification, chromatin remodeling, and non-coding RNAs [25].
Fig. 1
Illustration of epigenetic changes influenced by oxidative stress mediated hyperglycemia causing DN as an effect of metabolic memory Epigenetic changes in pathogenic genes linked to DN. Renal injury is caused by a variety of epigenetic changes that have been observed in protective genes, inflammatory and fibrotic genes, and even non-coding RNAs (ncRNAs) under diabetes circumstances. H3K4me1/2/3 mediated by histone methyltransferases like SET7/9 and H3K9/14ac, H3K18/23/27ac, and H4K5/8/14ac mediated by histone acetyl transferases like p300, WDR5, and PCAF are involved in the synthesis of active chromatin. On the other hand, protective gene transcription is suppressed by H3K9me2/3 and H3K27me3, which are mediated by HMTs EZH2 and SUV39H1, respectively, and histone PTMs mediated by HDACs Sirt6 and HDAC9. Transcriptional repression is also linked to DNA methylation that is facilitated by DNA methyltransferases. Dysregulation of ncRNAs, which participate in permanent epigenetic changes, can also result from epigenetic modifications in disease states including DN. The functions of epigenetic changes in metabolic memory are significant for pathogenic genes implicated with DN. HATs: histone acetyl transferases; HMTs, histone lysine methyltransferase; HDACs: histone deacetylase; PTMs, post-translational modifications
Histone modification involves its methylation, demethylation, and ubiquitination. Histone methylation is mostly associated with the N-terminus of lysine or arginine residues at the N-terminus of histone methyltransferases (HMTs) and histone demethylases (HDMs). Lysine residues can either be mono-methylated, di-methylated, or trimethylated; whereas, arginine residues can be mono-methylated, di-methylated asymmetrically, or symmetrically. Multiple sites of methylation have independent consequences. For instance, methylations associated with transcriptional activation have been found on histone H3 at lysine 4 (H3K4), arginine 17 (H3R17), H3K36, and H3K79 [26, 27]. Histone demethylation can be well understood by this illustration, which involves the role of angiotensinogen promoter in the renal proximal tubules of the diabetic model compared with that of the non-diabetic control model. This disclosed epigenetic modification is initiated from H3K9ac (Histone 3 lysine 9 residue acetylation) to H3K4me3 (Histone 3 methylation at 4 lysine residue), accompanied by demethylation at the departed instance of time, causing elevated gene expression [28]. Despite restoring glucose balance in the diabetic model, changes in DNA methylation and gene expression remained, indicating metabolic memory is mediated by cooperative epigenetic mechanisms. Histone modification controls various signaling pathways, and modifications in non-coding RNAs, such as miRNAs and lncRNAs, are linked to metabolic memory after glucose normalization [13, 25, 29].
Unlike histone modification, histone ubiquitination is regulated by ubiquitinating and deubiquitinating enzymes through the covalent binding of 76-amino acid proteins. It occurs at H1, H2A, H2B, H3, and H4, which are implicated in regulating the response to DNA damage, transcription, and genotoxic stress [30, 31]. Serine, threonine, and tyrosine histones are often phosphorylated at position H3 or H2A. This process is linked to centromere function, chromosomal condensation, and transcriptional activation. Enzymatic cascades, including ubiquitin ligases such as E1 activating enzymes, E2 conjugating enzymes, and E3 ubiquitin ligases, mediate the multistep process of ubiquitination. Research revealed that ubiquitination plays a major role in the development of DN [32]. The crucial phase in the activation of nuclear factor kappa B (NF-κB) is the ubiquitination of IκB and NF-κB dissociation, which is essential for the generation of inflammatory cytokines associated with diabetes mellitus. As was previously mentioned, ubiquitination plays a role in the development of DN by activating TGF-β and NF-κB by destroying the associated signal proteins [33]. The most recent research, conducted both in vitro and in vivo, revealed that the tripartite motif-containing E3 ubiquitin ligase, known as TRIP13, enhanced the ubiquitination and degradation of the C/EBP homologous protein, or CHOP, which is linked to renal injury. This, in turn, reduced the amount of collagen synthesis induced by DN and restored renal function. From these research findings, we could gain new insights into ubiquitin application as a therapeutic approach to DN. In order to prevent DN, further study is needed to uncover the histone ubiquitination’s hidden targets, according to the literature and experiments that are currently available [34].
Epigenetic regulation in metabolic memory is also assisted by studies using biological fluids from diabetes patients, primarily genomic DNA from archived whole blood or isolated white blood cells, which plays a predominant role in inflammation. There are also various studies revealing that gestational T1D and hyperglycemia in utero can affect renal function in offspring through DNA methylation, with differentially methylated CpGs associated with renal function [24]. Patients with poorly controlled T2D had chronic over expression of p66Shc (oxidative stress response protein). Another strategy for knocking out metabolic memory is to target non-coding RNAs (ncRNAs), namely ENST00000600527, NONHSAT037576.2, and NONHSAT135706.2 [35]. It has been observed that lncRNA E330013P06 in mice and MIR143HG in homo sapiens are activated in macrophages under diabetes circumstances targets metabolic memory. This lncRNA (MIR143HG) regulates the expression of macrophage inflammatory genes and foam cell formation, thus implicating lncRNA in DN progression. Furthermore, lncRNAs like plasmacytoma variant translocation 1 were recognized as potential cores for ESRD and are significantly associated with the pathogenesis of DN. Therefore, ncRNAs as epigenetic regulators can control gene expression through a variety of transcriptional and post-transcriptional processes, where they have attracted a lot of attention to their function in DM complications. This is because they can be used to fine-tune the responses to diabetogenic stimuli [36]. Researchers are still progressing their investigations on the significance of lncRNAs in human illness and developing strategies to use them therapeutically [29]. To target miRNAs and lncRNAs in this way, locked nucleic acid (LNA)-modified oligonucleotides are frequently used. The function of miRNA can be altered by LNA-modified anti-miRNA, antagomirs (miRNA inhibitors), or miRNA mimics. For instance, the LNA-modified inhibitor of miR-192 improved renal fibrosis and other DN-related symptoms in a diabetes-induced mouse model by reducing miR-192 levels and downstream miRNAs [37, 38]. Moreover, an LNA-modified antisense oligonucleotide called GapmeR, which was intended to target the DN-associated lncRNA megacluster, or lnc-MGC, was also successful in reducing the characteristics of early DN in mice and in human kidney cells. This GapmeR may be a way to stop the amplification circuits mediated by ER stress and chronic unfavorable events in the diabetic kidney. Other lncRNAs may be targeted by ncRNA targeting techniques in order to address metabolic memory [39]. Recent studies have shown that miRNAs and lncRNAs play significant roles in regulating gene expression and influencing the actions of growth factors and inflammatory factors associated with DN. Notably, the involvement of miRNAs in DN has been thoroughly investigated, particularly concerning TGF-β’s role in fibrosis and other crucial renal outcomes both in vitro and in vivo. Furthermore, advances in nucleotide chemistry and the development of nuclease-resistant anti-miRNAs have demonstrated effective in vivo targeting of miRNAs, highlighting the promising future potential of miRNA-based therapies for treating human diabetic complications [40]. Moreover, compared to protein-coding genes, most lncRNAs express themselves at significantly lower levels and have tissue-specific expression patterns. By serving as host genes for miRNA, attracting chromatin-modifying complexes to important gene loci, serving as sponges for miRNAs, and serving as scaffolds to bring protein complexes together, they can influence gene expression through a variety of epigenetic methods. Studies on in vitro and in vivo models of DN have revealed that after being removed from diabetic stimuli, variations in chromatin states and epigenetic histone PTMs at important pathological genes, as well as ncRNA expression, continue to play important roles in metabolic memory associated with an increased risk of DN [41]. However, as epigenetic modifications can be locus- and cell-specific, site-specific targeting is essential to reduce off-target effects. Through site-directed CRISPR-Cas9 editing, we can achieve this specificity by allowing for customized fusion structures, locus-specific changes in DNA methylation, or direct transcriptional regulation [42]. However, there remains much to discover about the scientific principles underlying each of these strategies, and they have great potential for use in clinical settings. In the clinic, the discipline of nucleotide-specific gene editing for rectifying genetic defects linked to disease is already showing promise. Epigenetic editing and epigenetic medications are likely to bring about translational changes in metabolic memory in the future [25].
Cellular mechanismStudies have shown that metabolic memory is influenced by biological processes like oxidative stress, non-enzymatic protein glycation, and inflammation. These events compound abnormal metabolism, leading to organ disease and cellular abnormalities. Oxidative stress and inflammation induce epigenetic alterations, activating genes linked to programmed cell death [19].
Brownlee et al.’s studies reveal that hyperglycemia leads to elevated reactive oxygen species production, distinguishing it from other clinical responses. This is due to increased electron donors in the tricarboxylic acid cycle, promoting excessive ROS and superoxide production [19, 43]. Elevated blood sugar levels increase diglyceride synthesis, activating protein kinase C and NADPH oxidase, leading to increased O2—production. This causes ROS to pass through cell membranes, damaging DNA and mitochondria, intensifying the accumulation of ROS, and perpetuating a harmful cycle. Even after the glycemic level returns to normal, hyperglycemic metabolic memory can be explained by a persistent overproduction of ROS [19, 44]. Interestingly, excess free ROS have a short half-life, but they remain in the circulation when blood glucose returns to normal, which plays a role in memory and cell damage. By interacting at the mitochondrial level, broad-band antioxidants can counteract these harmful effects of ROS. Furthermore, it has been noted that an overabundance of ROS can activate a variety of pathogenic cellular pathways, such as increased fluxes of polyol and hexosamine, AGEs, and NF-kB-induced vascular inflammation [19, 45].
AGEs are produced externally from outside sources as well as internally via a complicated “Maillard reaction,” which synthesizes a wide array of glycosylated adducts [46]. They mediate cytopathic alterations, insulin resistance, inflammation, and oxidative stress, and are higher in individuals with diabetes, MAFLD, and related comorbidities. These AGEs have also been connected to mortality and the chance of the illness progressing [47]. However, even after establishing glycemic control, diabetes patients’ blood vessels, kidneys, and hearts still contain proteins altered by AGEs because of their susceptibility to collapse. AGEs may therefore be significantly functional in metabolic memory.
Three basic molecular processes responsible for the harmful effects of AGEs include extracellular protein modification, internal protein modification, and signaling cascade activation by binding to the cell surface receptor for AGE (RAGE) [19]. The pathogenic mechanism involves AGE-to-RAGE binding, an immunoglobulin receptor. This interaction triggers complex signaling cascades, producing pro-inflammatory proteins and reactive oxygen species, promoting inflammation and tissue damage [48]. AGE-RAGE signaling initiates downstream pathways, resulting in over-expressed cytokines and adhesion molecules. A RAGE deficit can tend to cause inflammatory attenuation and immune cell recruitment inhibition. RAGE activation increases interferon-sensitive genes and JAK/STAT signaling activity [49]. Interactions between AGE and RAGE enhance inflammatory responses, leading to increased RAGE production. AGE-RAGE activation impairs mitochondrial function and raises oxidative stress, leading to vascular inflammation [50].
Metabolic memory and DNOne of the high-risk consequences of diabetes is diabetic nephropathy (DN). The patients subjected to DN have differential methylation of genes, including UNC13B, linked to hyperglycemia-induced apoptosis. This methylation, triggered by glucose stress, induces oxidative stress and apoptosis, contributing to DN. Upregulation of TGF-β1 improves Ras activation, promoting cell proliferation and fibrosis [51].
Research on histone modification of important metabolic genes revealed that epigenetic control plays a crucial role in the pathophysiology of DN. Histone variations impact oxidative stress, inflammation, and fibrosis in ways that contribute to the advancement of DN [52, 53]. For instance, diabetic glomerular thylakoid cells show altered histone methylation at various locations. These changes include a surge in H3K4me1/2/3, H3K36me2/3, and H3K79me2, which stimulate inflammatory responses and extracellular matrix accumulation, and a decrease in H3K9me2/3 and H3K27me3, which contribute to the inhibition of the renal fibrosis process [54]. Further, the development of DN mediated by renal inflammation is linked to histone changes. These alterations play a role in the activation of inflammatory factors such as NF-κB by macrophages and monocyte infiltration during hyperglycemia. Studies using epigenome profiling have shown differences in H3K4me2 and H3K9me2 for genes associated with inflammation and diabetes in monocytes exposed to hyperglycemia compared to normoglycemic controls [55]. T1DM patients’ blood monocytes and lymphocytes showed comparable epigenetic alterations. It has been discovered that HMT SET7 is a promoter of H3K4 methylation, which may co-activate pro-inflammatory genes in monocytes that are downstream of NF-κB. More research has been conducted on the relationship between DN and histone acetylation changes [56, 57]. In DN, the expression of p300, CBP, and other HATs is elevated, which leads to increased transcription of pro-inflammatory and pro-fibrotic proteins that worsen glomerular dysfunction linked to DN. H3K9/14Ac levels are successfully lowered by inhibition of p300/CBP, providing a possible therapeutic strategy for the management of hyperglycemia-mediated chronic kidney damage [58].
Additionally, ncRNAs have a crucial role as regulators in the development of DN. MiR-192, which binds TGF-β/Smad3 to up-regulate ECM and collagen as well as induce renal fibrosis, was initially discovered to be down-regulated in DN [59, 60]. It is thought that a number of miRNAs influence important aspects of DN, including apoptosis, fibrosis, and hypertrophy. It was shown that high glucose induction increased the expression of miR-21, miR-34a-5p, and miR217 in addition to promoting the activation of inflammatory pathways, inducing oxidative stress, and causing tissue fibrosis. 400, 401, and 402 LncRNAs have also been linked to DN in recent studies. It has been discovered that PVT1, a variation of the lncRNA plasmacytoma, has a role in the pathophysiology of DN and fibrosis. Increased pro-fibrotic element cellular expression in hypoglycemia is connected with glomerular thylakoid cell PVT1 up-regulation [61, 62].
Therapeutic interventionsSeveral environmental or lifestyle variables, such as obesity, high-fat meals, and physical inactivity, have a significant impact on how metabolic memory builds up. Lifestyle therapies, which include suitable dietary adjustments and exercise, have been shown to have significant positive effects on immune system function and metabolic balance [63]. These interventions may also help prevent or reduce the risk of metabolic illnesses. Recent research has shown how dietary changes might affect epigenetic changes, which in turn can interfere with potential long-term negative impacts mediated via metabolic memory [64]. Dietary bioactive substances like terpenoids and polyphenols operate as epigenetic modifiers, and if taken in the right amounts, they can undo epigenetic changes in genes linked to metabolism in children of low-nutrition mothers [65, 66].
Lack of exercise or a sedentary lifestyle is one of the primary risk factors for the development of illnesses associated with metabolic disorders. Exercise has been demonstrated in the past to help reduce insulin resistance, which has lately been linked to potential changes in epigenetic alterations [67]. Research has demonstrated that acute exercise causes a reduction in the promoter methylation levels of several genes, including PPARγ, MEF2, and peroxisome proliferator-activated receptor gamma co-activator 1 (PGC1), which in turn causes an increase in the expression of those genes. Insulin-resistant skeletal muscle has reduced PGC1, which triggers mitochondrial malfunction and raises intracellular lipid levels in myocytes, hence exacerbating insulin resistance [68]. By increasing PGC1 expression and activity in skeletal muscle, exercise could aid insulin resistance’s endurance and advancement. A six-month exercise intervention was shown to significantly modify the CpG methylation levels of numerous genes associated with diabetes and obesity in the adipose tissue of healthy people. These changes were supported by changes in mRNA expression [69]. Following sixteen weeks of aerobic training, skeletal muscle from sedentary patients with metabolic diseases also revealed hypomethylation of the promoter regions of genes including NRF1, solute carrier family 27 member 4 (SLC27A4), 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3), and GSK3A. This is thought to be related to the reduction of circulating lipids and improved glucose metabolism [70]. Furthermore, different forms of physical activity affect the expression of miRNA in both healthy individuals and those with metabolic diseases [
留言 (0)