HDR/Barakat syndrome is a rare autosomal dominant inherited condition manifesting with hypoparathyroidism, sensorineural deafness and/or renal dysfunction [5]. Here, we report on an infant with symptomatic hypocalcaemia, suppressed PTH, hearing and renal impairment as well as developmental delay and dysmorphism. There are diverse clinical features reported in 10p deletion syndromes ranging from dysmorphism to disparate cognitive, behavioural and developmental phenotypes. Moreover, 10p15 deletions are reported to be remarkably rare as an isolated finding; often complemented by duplication at a different location on the same chromosome, or a different chromosome altogether [6]. This was demonstrated in our case with both a terminal deletion and an interstitial duplication on the short arm of chromosome 10. Deletions of 10p15.3, the most distal segment of 10p, are associated with 10p15.3 microdeletion syndrome. Haploinsufficiency of ZMYND11 was identified as the underlying cause of the clinical phenotype of 10p15.3 microdeletion syndrome. This syndrome is associated with developmental delay, characteristic dysmorphic features, behavioural disturbances, hypotonia, seizures, low birth weight, short stature, genitourinary malformations and recurrent infections [7].
GATA3 is a transcription factor consisting of conserved binary zinc-finger domains which plays an integral role in the development of parathyroid glands, kidneys and the auditory system, including the inner ears [8]. GATA3, the causal link to HDR syndrome, maps to 10p14-15 [9]. Two patients with Barakat syndrome were identified by deletion mapping studies which identified the critical 200-kb region that comprises GATA3 [3]. CMA is a high-resolution technique capable of detecting chromosomal aberrations as small as 50 kb. It is a relatively affordable diagnostic tool for whole genome analysis and is the recommended first-tier test for the investigation of suspected microdeletion/duplication syndromes, neurodevelopmental delay and intellectual disability [10]. The analysis is superior to conventional cytogenetics, making it valuable in low-resource settings with limited access to advanced diagnostic technologies [11, 12]. This is a rare case of HDR syndrome diagnosed on CMA as a 10p15 deletion; and to our knowledge, is the first published case of an African patient. We know of two case reports of HDR syndrome diagnosed by CMA; the one patient had monosomy of 10p15.3p13, while the other had a pathogenic deletion of 10p15.3p14 [13, 14]. The 10p deletion was associated with duplication of different chromosomes, namely 20p13p12.3 and 12p13.33p13.33, respectively. In addition to the clinical features of Barakat syndrome, these patients presented with dysmorphic features and developmental delay resembling our case presentation.
Cases of inverted duplication and deletion of chromosome 10p have been rarely reported in the literature and differ with regard to chromosome breakpoints [15, 16]. Chromosome rearrangements resulting in a terminal deletion and adjacent inverted interstitial duplication have been reported to occur on most chromosomes. The most common mechanism resulting in this rearrangement is U-type exchange between sister chromatids. Recombination events resulting from the presence of inverted low copy repeats, or having one parent carry a paracentric inversion, can also lead to this type of rearrangement. The latter two mechanisms typically result in a region of normal copy number between the deletion and inverted duplication. High resolution microarray with sufficient probe coverage can be used to distinguish between these mechanisms [17]. This is important to clarify recurrence risks for future pregnancies.
The interstitial gain of chromosome 10p14p13 with CNV size of approximately 4.4 Mb detected in our case is noteworthy. One patient with a duplication in this region presented with autistic features and global developmental delay (DECIPHER; 301137).
Bernandini et al. reported a case of HDR syndrome with 10p15.3p15.1 deletion as well as 10p15.1p14 duplication. Interestingly, follow up with a real-time PCR assay confirmed the increased GATA3 gene expression leading to Barakat syndrome [16]. Kim et al. published a neonate with the largest deletion reported to date, spanning 16 Mb on chromosome 10p. As expected, the clinical presentation was extensive, with major congenital abnormalities and features of HDR syndrome [18]. The two published cases are presented in Table 2 alongside the present case, highlighting shared clinical features across the three patients. Differences in phenotype may be ascribed to the varying sizes of the CNVs in each patient and their precise breakpoints.
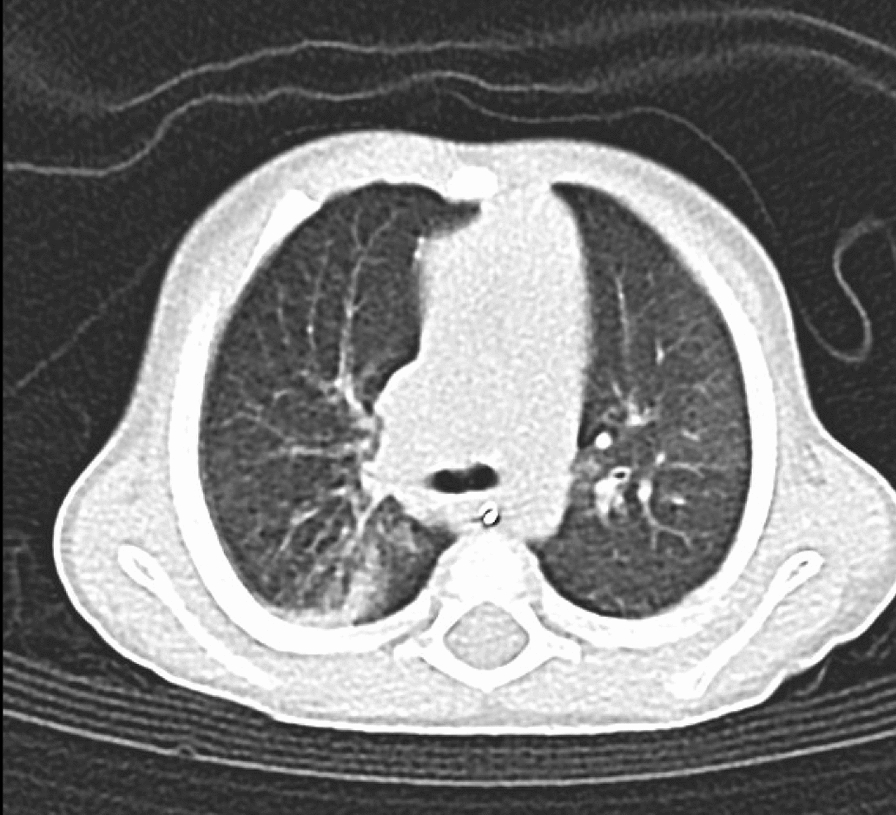
Table 2 Comparison of the patient’s phenotype with the two published cases with inverted duplication and deletion of chromosome 10pHDR syndrome is marked by a genetic and clinical heterogeneity with only some clinical manifestations notable in affected patients. This remarkable heterogeneity is attributed to the incomplete penetrance and variable expression as it pertains to the age of onset, severity and extent of the manifested abnormalities. Bilateral sensorineural deafness is reported as the most consistent symptom that is encountered early in the disease, while the presence of hypoparathyroidism and renal diseases is highly variable [19]. The renal complications may be subtle and functional with no renal structural abnormalities, as demonstrated by mild renal impairment and proteinuria in our patient. The renal structural abnormalities are variable and may be asymmetrical, ranging from hypoplastic to cystic kidneys and pelvicalyceal anomaly. The mild renal involvement may infer favourable outcomes for this patient because the prognosis of HDR is dependent on the severity of the kidney disease.
As demonstrated in our case, severe hypocalcaemia with seizures is often the reason that affected patients present to health care facilities for urgent medical care. Initially, the refractory hypocalcaemia could not be corrected with intravenous and oral calcium along with active vitamin D supplementation. Recombinant PTH replacement therapy is recommended in instances of unresponsive hypocalcaemia [20]; this treatment option was not available in our setting. There are no current guidelines on long-term surveillance of patients; however, frequent serum calcium monitoring is recommended until stabilized, to be followed by 2–3 monthly monitoring as an outpatient. Close monitoring of the renal function test is paramount in anticipation of nephrocalcinosis due to calcium over-supplementation [21]. Genetic counselling is a critical part of the patient’s management and should be considered routine care for patients diagnosed with chromosomal anomalies. Genetic counsellors can explain the diagnosis to parents and families, discuss recurrence risks and further genetic testing recommendations and provide psychosocial support to families.





留言 (0)