
記住我




A549 (ATCC, USA, CRM-CCL-185): the human lung adenocarcinoma epithelial cell line was obtained from the American Type Culture Collection, Virginia, USA. Cells were grown in MEM GlutaMAX (Gibco, USA, 41,090–028) supplemented with 10% fetal calf serum (FCS) GOOD, (PAN Biotech, Germany, P40-37500) and 1% penicillin–streptomycin, (PAN Biotech, Germany, P06-07100).
THP-1 (DSMZ, Germany, ACC 16): the human monocytic leukemia cell line was obtained from the German Collection of Microorganisms and Cell Cultures GmbH, Braunschweig, Germany. Cells were grown in Roswell Park Memorial Institute medium (RPMI 1640), (PAN Biotech, Germany, P04-17500) supplemented with 10% FCS GOOD, 1% penicillin–streptomycin, 1% L-Glutamine (PAN Biotech, Germany, P04-80100)., 10 mM HEPES buffer (PAN Biotech, Germany, P05-01100) and 1 mM sodium pyruvate (PAN Biotech, Germany, P04-43100).
THP-1 differentiation: THP-1 cells were differentiated to macrophages for 48 h with 100 nM of PMA (Sigma Aldrich, Germany, P1585) in RPMI medium. Following this 48-h period, PMA containing medium was removed and the cells was replenished with fresh RPMI medium.
NR8383 (ATCC, USA, AgC11 × 3A, NR8383.1): rat macrophage cell line originally isolated from the lungs of a normal rat, were obtained from the American Type Culture Collection, Virginia, USA. Cells were grown in Ham’s F-12 K (Kaighn’s) Medium (Gibco, USA, 21,127,022), supplemented with 15% FCS GOOD and 1% penicillin–streptomycin.
After thawing from cryopreservation each cell line was kept in cell culture for a total of four passages, to allow for a degree of phenotypical stabilization. Cell lines were then passaged twice per week, every 3 or 4 days. All cell lines and were incubated at 37 °C, 5% CO2. Cell cultures underwent routine testing for mycoplasma contamination, consistently yielding negative results throughout the experimental period.
Cell culture setupsIn a first step, we evaluated the effects of culturing cells of three different cell lines in different cell culture vessels, i.e., 6WP-wells and 10 cm dishes. To address discrepancies between the recommended cell numbers and cell culture media (CCM) volumes by cell line providers, we used cell numbers and CCM volumes that align with standard recommendations, which are also commonly utilized in scientific literature. An overview of these conditions is listed in Tables 1 and 2. The cell density was kept constant for both cell culture vessels (Table 1) and is calculated considering cell number divided by vessel’s area. Due to the different CCM volumes used, 6WP-wells had higher volume to surface area ratio (mL/cm2) and CCM filling height compared to the 10 cm dishes (Table 2). Thus, these represent key differences of the experimental setup, which were assessed. 24 h after seeding the cells in the corresponding vessels the cells were harvested and processed for proteomic analysis.
Table 1 Comparison of the cell seeding densities for the different cell lines in both culture vesselsTable 2 Metrics for each vessel comparing the different parametersCells were seeded as detailed in Table 1, across three biological replicates derived from distinct cell passage numbers. For each biological replicate, we performed three technical replicates, using cells from the same passage number but grown in separate culture vessels. The cell density was maintained constant across different vessels relative to their surface area.
The characteristics of the cell culture vessels used for the experiments are presented in Table 2.
The cell passage number rangeIn a second step, we investigated alterations in the proteomes of A549, dTHP-1, and NR8383 cells focusing on variations across different subcultures of the same stock. The cells of different passage number were grown in 10-cm dishes or 6WP-wells and harvested 24 h after cell seeding. The cultivation times were adapted to fit the practical constraints of the laboratory schedule, ensuring that cell splitting could be integrated smoothly into the regular workweek. Specifically, A549 cells for five passages (2.5 weeks), and dTHP-1 cells for seven passages (3.5 weeks) and NR8383 cells were cultured for up to three passages (equivalent to 1.5 weeks). This approach reflects a realistic scenario in toxicological research, where experiments must often be designed to accommodate the standard workweek, thereby ensuring to obtain three biological replicates for robust toxicological testing.
Differentiation process of THP-1 cellsIn the third step, we investigated the proteomic changes occurring in dTHP-1 cells following the completion of the PMA-induced differentiation process. Innately monocytic cells, THP-1 acquire a macrophage-like phenotype and functional state through differentiation induced by PMA, a compound from the phorbol ester family. However, a notable limitation of differentiating THP-1 cells by PMA is that they enter an (pro-)inflammatory state, a change anticipated to be reflected in their proteomic profile. Following the standard procedure for THP-1 cell differentiation (Cam and de Mejia 2012; De et al. 2021), after the 48-h differentiation period with PMA, the PMA-containing medium was removed and the cells were supplemented with PMA-free CCM. Cells were then harvested at three distinct time points: 6, 24 and 48 h post-PMA treatment.
MS sample preparation, liquid chromatography–electrospray ionization–tandem mass spectrometry (LC–ESI–MS/MS) measurementsCell pellets were harvested, lysed and digested for proteomics measurements using the iST kit (PreOmics, Germany, P.O.00030). Desalted peptides were reconstituted in 0.1% (v/v) TFA, 5% (v/v) acetonitrile to a final concentration of 50 ng/µl and transferred to vials with glass inserts. LC–MS analyses were performed on an UltiMate 3000 RLSCnano system (Thermo Scientific, USA) connected to an Orbitrap QExactivePlus (Thermo Scientific, USA) mass spectrometer.
The LC system was coupled to the mass spectrometer via a nanospray flex ion source equipped with a stainless-steel emitter (Thermo Scientific, USA). Samples were injected (250 µg) and concentrated on an Acclaim PepMap100 C18 trap column (3 μm, 100 Å, 75 μm i.d. × 2 cm, Thermo Scientific) equilibrated (5 µL/min, 5 min, 45 °C) with 0.05% TFA, 2% acetonitrile in water. After switching the trap column inline, peptides were separated on an Acclaim PepMap100 C18 column (2 μm, 100 Å, 75 μm i.d. × 25 cm, Thermo Scientific) at an eluent flow rate of 0.3 µL/min using a two linear gradient (5 to 35% B in 90 min, 35 to 50% in 5 min). Mobile phase A contained 0.1% formic acid in water, and mobile phase B contained 0.1% formic acid in 80% acetonitrile. Non-targeted analysis was performed in a data-dependent acquisition (DDA) mode, fragmenting the ten most abundant, multiply charged ions, with dynamic exclusion time set to 60 s. Each sample was measured in three analytical replicates. A full list of instrument parameters is given in Supplementary Table 1.
Protein identification and data analysisMass spectrometric data from each LC–MS run were analyzed using the MaxQuant software (Cox and Mann 2008; Tyanova et al. 2016) (Version 1.6.14). The identification of proteins was performed using the MaxQuant-implemented Andromeda search engine against a reference Homo sapiens proteome. Precursor and fragment mass tolerance were set to 7 ppm and 0.5 Da, respectively. Variable (methionine oxidation and N-terminal acetylation) and fixed modifications (cysteine carbamidomethylation) were set for the search and trypsin with a maximum of three missed cleavages was chosen for searching. The minimum peptide length was set to seven amino acids and false discovery rate (FDR) for peptide and protein identification was set to 0.01. Proteins were quantified via label-free quantification (LFQ) as detailed in Stobernack et al. 2024 (Stobernack et al. 2024).
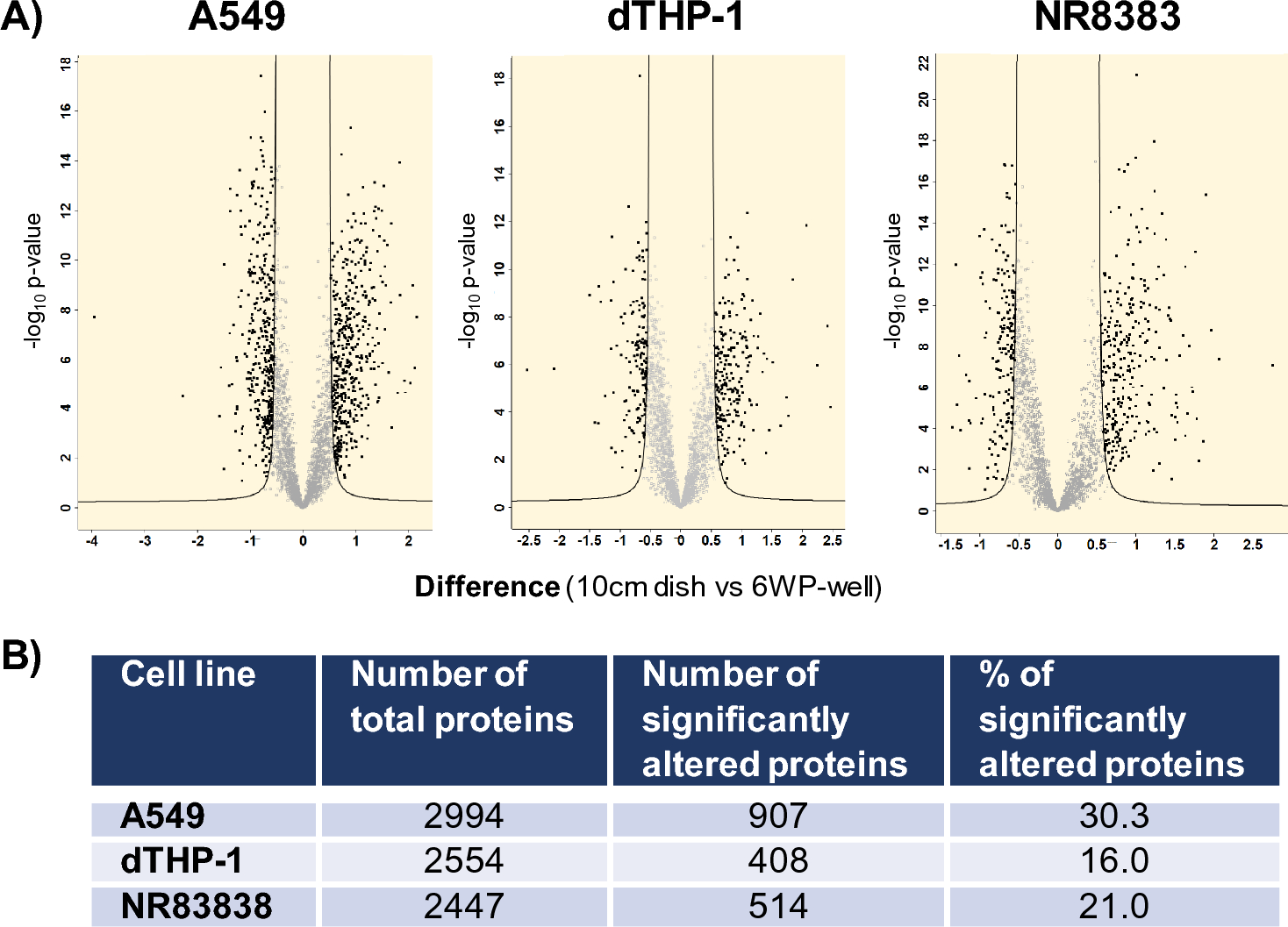
Statistical analysisAll conducted experiments included three biological replicates for each sample and were analyzed in three technical replicates. In this study, protein LFQ intensities were pre-processed by log2 transformation. The Perseus software (Tyanova et al. 2016) was utilized to identify proteins with significant alterations between two compared conditions or groups. We identified significantly altered proteins using FDR settings of 0.01 and s0 set at 0.1, as employed in multi-volcano analysis, based on criteria established by Rudolph et al., 2019 (Rudolph and Cox 2019). The software provides an analysis summary in a data matrix format, which includes t-test significance, -Log t-test values, p-values, and t-test differences. Volcano plots, as shown in Fig. 1, graphically represent the significance and magnitude of changes in protein expression, combining the functions of the two-sample t-test and the scatter plot. Significantly altered proteins were then subjected to KEGG pathway analysis using the R package enrichR (Kuleshov et al. 2016). The AnnotationDBI package was used to convert Uniprot IDs to Entrez IDs. Prism 9.3.1 (GraphPad Software, San Diego, CA, USA) and Excel (Office Professional 2021 Plus, Microsoft, Redmond, USA) were used for calculations, filtering, and graphical display.
Fig. 1
Effect of the cell culture setup at the proteome level assessed 24 h after cell seeding A Volcano plots depicting the impact of the cell culture setup selection (10 cm dishes vs. 6WP-wells) on the proteome profiles of the different cell lines: A549, dTHP-1 and NR8383. B Total proteins detected per cell line and proteins significantly altered upon seeding in different vessels, presented as both the total count and percentage
留言 (0)