
記住我

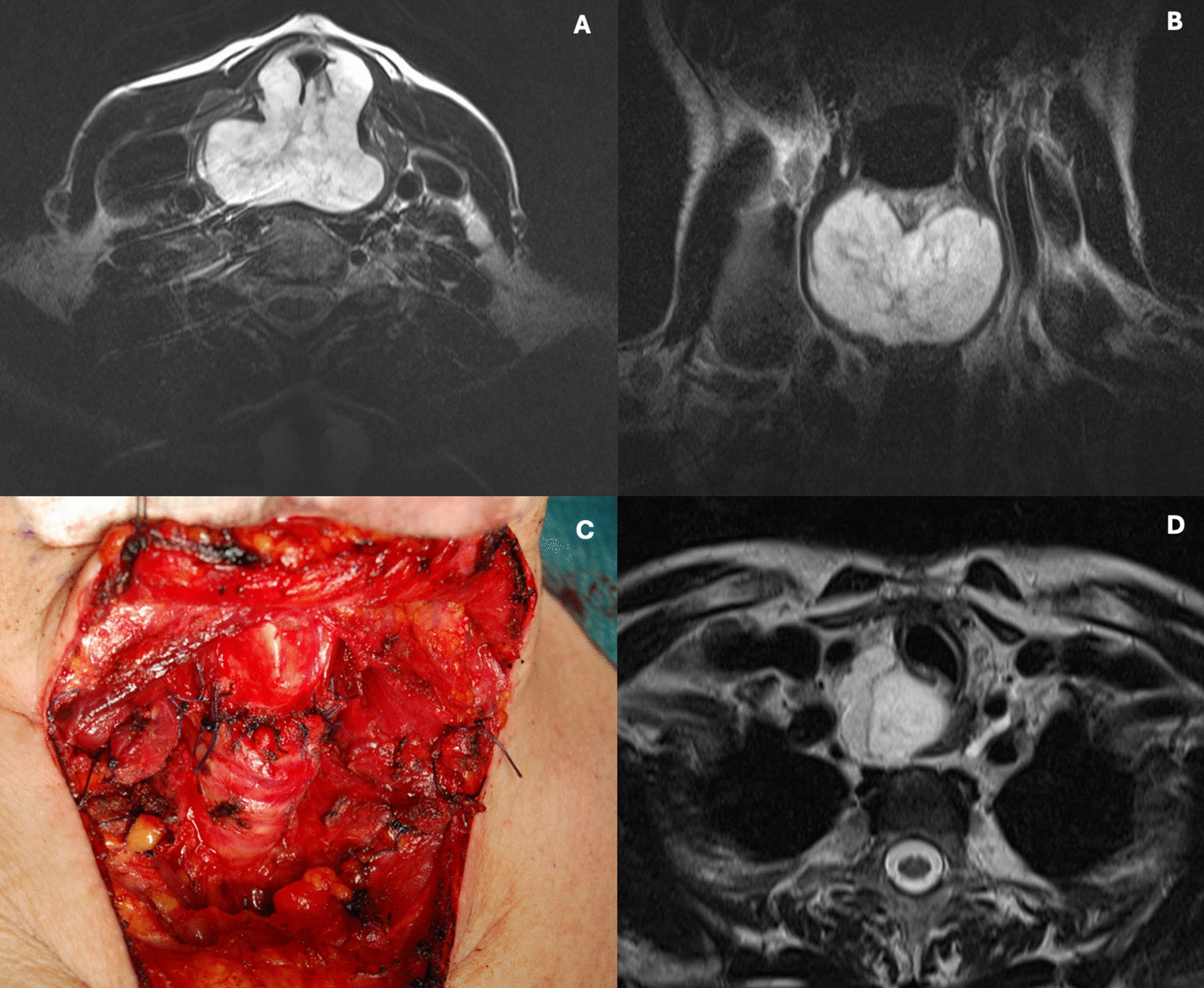

The multicenter, open-label, single-arm, phase I/II CA209-9KH clinical trial (registration numbers: EudraCT 2017-003404-52; ClinicalTrials.gov NCT04098432) was conducted to investigate the safety and tolerability of stereotactic radiotherapy followed by nivolumab in patients with locally unresectable non-metastatic PDAC, and the anticancer activity of nivolumab after previous stereotactic radiotherapy.
The trial enrolled patients from three cancer centers, and included those aged ≥ 18 years, with histologically confirmed, unresectable non-metastatic PDAC after four cycles of induction FOLFIRINOX chemotherapy and who signed informed consent to participate. The tumor had to be considered unresectable after induction chemotherapy and without evidence of disease progression (Fig. 1). Tumor samples from all enrolled patients were histologically confirmed and centrally reviewed. Other inclusion criteria included measurable disease as per RECIST 1.1 and iRECIST criteria, Eastern Cooperative Oncology Group (ECOG) Performance Status 0 or 1, acceptable laboratory parameters (aspartate aminotransferase ≤ 3 × upper limit of normal (ULN), alanine aminotransferase ≤ 3 × ULN, total bilirubin ≤ 1.5 × ULN (except patients with Gilbert syndrome who must have total bilirubin ≤ 3 × ULN), serum creatinine ≤ 1.5 ULN or creatinine clearance > 50 ml/min using the Cockcroft–Gault formula, white blood cells ≥ 2000/μl, neutrophils ≥ 1 500/μl, platelets ≥ 100,000/μl, and hemoglobin ≥ 90.0 g/l. The principal exclusion criteria included resectable disease, localization or size of the tumor that did not allow SRT, the presence of distant metastases, other previous antitumor therapy except the four cycles of induction FOLFIRINOX, previous therapy for other malignant disease within 5 years before trial inclusion (except epithelial skin tumors), presence of another synchronous malignant tumor, previous radiotherapy to the abdominal region, previous immunological treatment (anti-CTLA-4, anti-PD-1, or anti-PD-L1), active, known, or suspected severe autoimmune disease, major surgery less than 28 days before the first dose of study treatment, treatment with any investigational medicinal product within 4 weeks before trial enrollment, any positive test for hepatitis B or C virus indicating acute or chronic infection, and known history of testing positive for human immunodeficiency virus or known acquired immunodeficiency syndrome. SRT and immunotherapy were administered sequentially.
Fig. 1 Stereotactic Radiotherapy
Stereotactic RadiotherapyThe gross tumor volume (GTV) was defined based on planning computed tomography and magnetic resonance imaging. Only the primary tumor was delineated as the GTV (not a pathological lymph node). The clinical target volume (CTV) was not defined. The planning target volume (PTV) encompassed the GTV with a margin of 3 mm. The at-risk organs contoured included the left and right kidneys, liver, stomach, duodenum, small intestine, and spinal cord.
The prescribed dose was 32 Gy in four fractions within 4 weeks (8 Gy per fraction), as we hypothesized that irradiation with large fractions at weekly intervals might be most beneficial for the activation of antitumor immunity. The near-maximum dose within the PTV was less than 110% of the prescribed dose. The near-minimum dose in the PTV (D98) was greater than 85% of the prescribed dose. Plans had to be normalized such that 95% of the PTV had to receive at least 95% of the prescribed dose. The recommended dose was normalized when the prescribed dose corresponded to the PTV Dmean. The required tolerance doses for at-risk organs were consistent with the QUANTEC (Quantitative Analysis of Normal Tissue Effects in the Clinic) recommendations [23].
SRT was delivered using a linear accelerator. Three-dimensional (3D) conformal SRT and intensity-modulated radiotherapy techniques were used in this study. Image guidance using a cone beam was required before each radiation fraction. The first fraction of SRT was performed 4 weeks after the completion of induction chemotherapy.
Nivolumab TherapyNivolumab was started in the fourth week after completion of radiotherapy in a dose of 3 mg/kg intravenously every 2 weeks until disease progression or unacceptable toxicity. In cases of progression during immunotherapy, standard systemic chemotherapy (i.e., FOLFIRINOX, gemcitabine, or other regimens) or best supportive care was administered. Post-treatment follow-up according to the study protocol was indicated in cases with no progression during the clinical trial.
OutcomesThe primary endpoint of treatment safety was assessed as the incidence of adverse events (AEs) and serious adverse events (SAEs) according to the Food and Drug Administration (FDA) definition, deaths, and laboratory abnormalities. All events were reported, classified, and graded using the National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE) v4.03 criteria [24].
The following secondary objectives were evaluated: progression-free survival (PFS) and OS, the relationship between selected laboratory (CA 19-9 serum concentration) and histopathological (PD-L1 expression, mismatch repair proteins) markers and progression status, and the patients’ capacity to perform activities of daily living and QoL using the European Organisation for Research and Treatment of Cancer (EORTC) Core Quality of Life Questionnaire (QLQ-C30).
Complete medical history and demographics were collected at the screening visit. Physical examination, assessment of signs and symptoms, ECOG performance status, concomitant medication review and hematology test (blood count: red blood cells count, hemoglobin, white blood cells count and differential, platelets count) were done every visit (at the screening visit, weekly during radiotherapy, prior to initiation of nivolumab therapy, and every 2 weeks during nivolumab therapy and at the end of the treatment visit). Biochemistry test (sodium, potassium, calcium, ionized calcium, phosphate, magnesium, urea, creatinine, lactate dehydrogenase, conjugated and total bilirubin, total protein, albumin, aspartate aminotransferase, alanine aminotransferase, gamma-glutamyl transferase, alkaline phosphatase, total bilirubin, conjugated bilirubin, total protein, albumin, fasting glucose, amylase, C-reactive protein;) was performed at the screening visit, every 2 weeks during radiotherapy, prior to initiation of nivolumab therapy, and every 2 weeks during nivolumab therapy, and at the end of the treatment visit. Thyroid function tests (thyroid-stimulating hormone, free T3 and free T4) and urinalysis test were performed at the screening visit, prior to initiation of nivolumab therapy, every 4 weeks during nivolumab therapy, and at the end of the treatment visit. Imaging (computed tomography of pelvis, abdomen, and chest) and laboratory marker CA 19-9 as well as EORTC QLQ-C30 were completed at the screening visit, prior to initiation of nivolumab therapy, every 4 weeks during nivolumab therapy, and at the end of the treatment (EOT) visit. Tumor response was assessed by computed tomography performed every 8 weeks according to guidelines for response criteria for use in trials testing immunotherapeutics (iRECIST) [25].
Detection of PD-L1 expression was performed using 22C3 primary antibody (DAKO, Glostrup, Denmark) and a Ventana Optiview detection kit on Ventana Benchmark Ultra immunostainer. The expression was evaluated in a light microscope using the tumor proportion score (TPS), quantifying the percentage of neoplastic cells showing any membranous positivity (out of all neoplastic cells visible in the slide). The presence of mismatch repair proteins has been tested immunohistochemically using primary mouse antibodies against MLH1 (Zytomed, clone G168-15), MSH2 (Cell Marque, clone G219-1129), MSH6 (BioSB, clone EP49) and PMS2 (Ventana, clone A16-4), using the Ventana Optiview detection kit (for MLH1, MSH2 and PMS2, all on Ventana Benchmark Ultra immunostainer) and DAKO detection kit (on DAKO Omnis immunostainer).
Statistical AnalysisThe CA209-9KH clinical trial was exploratory and not powered to formally test any null hypotheses; therefore, no sample size calculation was performed. The study was planned to enroll 15–20 patients. Descriptive statistics for demographics and other baseline characteristics were used, including mean, median, range, and standard deviations (SD) for continuous data and absolute and relative frequencies for categorical data. The safety and toxicity profiles of the study treatments were assessed as the incidence of AEs, SAEs, deaths, and laboratory abnormalities.
Time-to-event endpoints were estimated using the Kaplan–Meier method. The median survival and 1- and 2-year survival rates were also calculated. The point estimates and the 95% confidence intervals are presented. The start date was the date on which the participants entered the study. Patients with no events were censored at study discontinuation.
The influence of PD-L1 and mismatch repair proteins expression on PFS was planned to test using log-rank analysis and Cox regression analysis. The median value was planned as the threshold for dividing into PD-L1 high and PD-L1 low groups. A two-sample t-test was used to compare QoL scores and CA 19-9 serum concentrations at treatment phase visits and at the EOT visit compared with values at the screening visit, with statistical significance set at p < 0.05.
Ethical StatementThe study protocol, informed consent forms for patients, and all other relevant documents were approved by the Ethics Committee of the University Hospital Hradec Králové (EC UHHK) and the local ethics committees of other participating centers: Ethics Committee of the University Hospital Olomouc and Ethics Committee of the Thomayer University Hospital. The EC UHHK has the approval of the Ministry of Health of the Czech Republic for multicentric clinical trials and is accredited in the USA by the Office for Human Research Protections (number IORG0008813).
The trial was conducted in accordance with the Guideline for Good Clinical Practice and the Declaration of Helsinki. All study participants were fully informed and signed informed consent forms to participate the study.
留言 (0)