In the era of precision medicine, NGS systems have radically modified the diagnostic paradigm for the clinical management of patients with solid tumors, allowing for more thorough mutational profiles and accurate test results than conventional approaches. Amazingly, these panels can cover between 2 and > 500 clinically relevant genes. However, interpreting the vast range of test results generated by NGS panels is a dauting (but nonetheless necessary) task for oncologists [24]. Indeed, identifying treatable concomitant pathogenetic/activating mutations drastically impacts the clinical management of patients with tumors [25]. By the same token, identifying “unknown” molecular alterations is equally paramount as it allows us to expand the molecular and clinical landscape of informative variants in patients with solid tumors [26, 27]. In recent years, having acknowledged the practical pitfalls in the clinical reporting of molecular alterations from routine diagnostic specimens, international societies have underlined the pivotal role of data interpretation systems in overcoming the challenges of interpreting NGS data [25, 27]. Among these systems are bioinformatics pipelines. Generally, bioinformatics pipelines allow for the automated clinical interpretation of detected variants based on dedicated clinical databases. Today, a rapidly growing number of technical solutions include cloud-based systems that can be either integrated or not into cloud-based platforms. Among the currently available tools for the molecular and clinical interpretation of NGS data are open-source NGS tools, commercial NGS tools, and alignment tools. An advantage of these NGS bioinformatics tools is that they can automatically perform three fundamental analyses, thereby reducing the overall working time. The primary analysis consists of base calling and scoring base quality through FASTQ or uBAM files; the secondary analysis involves read alignment or de novo assembly; finally, the tertiary analysis involves data annotation by means of the SIFT, PolyPhen-2 CADD, ANNOVAR, and Condel prediction tools.
The clinical interpretation of molecular variants, which are ranked on the basis of their pathogenicities, is assessed through several approaches. Among these approaches is PHIVE. PHIVE investigates the clinical actionability of molecular variants by comparing human disease phenotypes with data from knockout animal models. Other approaches include VarSeq/VSClinical (Golden Helix), ingenuity variant analysis (Qiagen), and the use of Alamut® software (interactive biosoftware) or VarElect software. These bioinformatics tools provide a clinical data repository of pathogenic alterations across solid tumors, hence representing a user-friendly, time-saving, and cost-effective diagnostic tool in clinical practice.
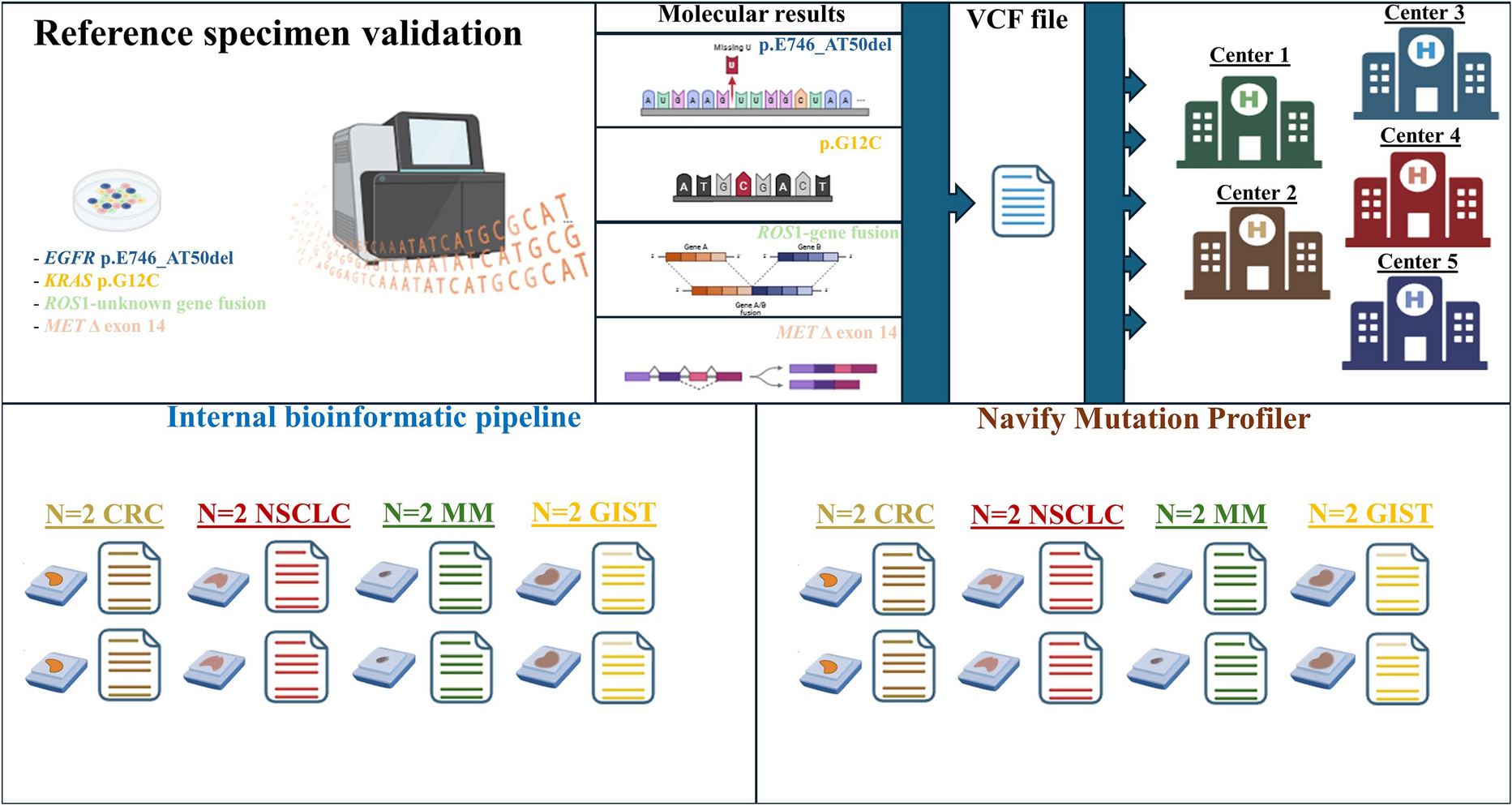
The downside of such heterogeneous landscape of databases is the lack of standardized interpretation workflows. Indeed, disharmonized parameters (in terms of record availability, clinical actionability, and interpretation systems) may often negatively affect the clinical interpretation of molecular records and, thus, the clinical outcomes of patients [28, 29]. A failure to detect or to accurately interpret tumor biomarkers is the reason why too many people are molecularly misdiagnosed, thereby missing the opportunity to benefit from targeted therapies [28]. In this paper, we evaluated the technical and clinical performance of Navify® Mutation Profiler—a tertiary genomic data analysis software that generates an automated tiered interpretation of molecular alterations through VCF files. Notably, built on publicly available evidence tiers, nMP automatically inspects molecular records by consulting multiple databases simultaneously [21]. Moreover, it integrates recommendations and updates on drugs approved by different agencies, including the European Medicines Agency, SwissMedic, and the National Institute for Health and Care Excellence [21]. Of note, the nMP system also allows “missed variants” to be reinterpreted manually, thereby assigning a tiered clinical interpretation of these variants in the final report.
In this study, we investigated the technical performance of the nMP software in interpreting and reporting NGS data from solid tumor samples. For this purpose, we used standard reference specimens [22] covering four different types of NSCLC actionable alterations (point mutation, deletion, aberrant rearrangement, and exon skipping mutation).
All involved institutions successfully carried out the molecular analysis. Before the interpretation of real-world samples, a reference VCF file was shared with the participating institutions. In this training session, high concordance rates were observed between the nMP software and the institutions’ bioinformatics pipelines for DNA-based alterations; however, in one institution (center 5), nMP failed to detect RNA-based alterations in a single case. A possible reason for the discrepancy between center 5 and the other institutions is that center 5 used the first version of the nMP software to analyze the reference VCF file; instead, the other institutions adopted a more recent bioinformatics pipeline (Oncomine_Fusions_only) optimized for the interpretation of RNA-based alterations (Supplementary File 1). In detail, the nMP software efficiently interpreted DNA and RNA molecular alterations, as evidenced by its overall concordance rates with the internal workflows, namely, 91.7% (44 out of 48) and 91.7% (11 out of 12), respectively. Moreover, a high percentage agreement of 85.7% (6 out of 7) was also seen between the nMP software and standard bioinformatics pipelines on the molecular interpretation of a wild-type series according to the referral interpretation approach. Regarding the discordant cases, in a single case, only the nMP system highlighted concomitant clinically relevant alterations in key genes for the clinical administration of patients with solid tumors (KRAS p.G12V plus NRAS p.G13D, center 2). We speculate that this discrepancy was due to the fact that the internal workflow systems filtered out the p.G13D hotspot mutation because the VAF threshold value was below the clinical value of 5.0% that is generally established for the interpretation of variants in tissue samples (Table 3). Conversely, the nMP system failed to detect two c-KIT pathogenetic duplications (c-KIT p.A502_Y503dup and p.T574_W577dup) in patients with GIST. The literature demonstrates that the detection of short modifications of repetitive DNA sequences requires optimized bioinformatics pipelines that are able to recover filtered-out variants from final reports [30]. Of note, the nMP software detected p.V600E BRAF mutations in a single instance (colon 1, center 5). By contrast, although the same BRAF mutation was identified by the internal workflow, it was not included in the final report out of clinical request (Table 3).



留言 (0)