Materials
All test substances were purchased in analytical grade (purity ≥ 98.0%) from Sigma-Aldrich, Pestanal® (Taufkirchen, Germany): Cyproconazole, CAS no. 94361–06-5, catalog no. 46068, batch no. BCCD4066; Fluxapyroxad, CAS no. 907204–31-3, catalog no. 37047, batch no. BCCF6749; Azoxystrobin, CAS no. 131860–33-8, catalog no. 31697, batch no. BCCF6593; Chlorotoluron, CAS no. 15545–48-9, catalog no. 45400, batch no. BCBW1414; Thiabendazole, CAS no. 148–79-8, catalog no. 45684, batch no. BCBV5436; 2-Phenylphenol, CAS no. 90–43-7, catalog no. 45529, batch no. BCCF1784. William’s E medium, fetal calf serum (FCS) good forte (catalog no. P40-47500, batch no. P131102), recombinant human insulin and l-glutamine were acquired from PAN-Biotech GmbH (Aidenbach, Germany), FCS superior (catalog no. S0615, batch no. 0001659021) from Bio&Sell (Feucht bei Nürnberg, Germany). Dimethyl sulfoxide (DMSO, purity ≥ 99.8%), hydrocortisone-hemisuccinate (HC/HS), hydrocortisone, epidermal growth factor (EGF) and neutral red (NR) were purchased from Sigma-Aldrich (Taufkirchen, Germany). Dulbecco’s modified eagle medium (DMEM) and Ham’s F Nutrition mix were obtained from Gibco® Life Technologies (Karlsruhe, Germany), trypsin–EDTA, Penicillin–Streptomycin and insulin-transferrin-selenium from Capricorn Scientific GmbH (Ebsdorfergrund, Germany).
Cell cultureHepaRG
HepaRG cells were obtained from Biopredic International (Sant Grégoire, France) and kept in 75 cm2 flasks under humid conditions at 37 °C and 5% CO2. Cells were grown in proliferation medium consisting of William’s E medium with 2 mM l-glutamine, supplemented with 10% FCS good forte, 100 U mL−1 penicillin, 100 µg mL−1 streptomycin, 0.05% human insulin and 50 µM HC/HS for 2 weeks. Then, HepaRG cells were passaged using trypsin–EDTA solution and seeded in 75 cm2 flasks, 6-well, 12-well and 96-well plates at a density of 20 000 cells per cm2. Cells in cell culture dishes were maintained in proliferation medium for another 2 weeks before the medium was changed to differentiation medium (i.e., proliferation medium supplemented by 1.7% DMSO) and cells were cultured for another 2 weeks. Thereafter, cells were used in experiments within 4 weeks, while media was changed to treatment media (i.e., proliferation media supplemented by 0.5% DMSO and 2% FCS) 2 days prior to the experiments.
RPTEC
The RPTEC cell line was obtained from Evercyte GmbH (Vienna, Austria) and cultivated as previously described (Aschauer et al. 2013; Wieser et al. 2008). Cells were grown in a 1:1 mixture of DMEM and Ham’s F-12 Nutrient Mix, supplemented with 2.5% FCS superior, 100 U mL−1 penicillin, 100 µg mL−1 streptomycin, 2 mM l-glutamine, 36 ng mL−1 hydrocortisone, 10 ng mL−1 EGF, 5 µg mL−1 insulin, 5 µg mL−1 transferrin and 5 ng mL−1 selenium. RPTEC were cultivated in 75 cm2 flasks until they reached near confluence. Then, cells were passaged using trypsin–EDTA and seeded at 30% density in 75 cm2 flasks for further sub-cultivation and 6-well, 12-well and 96-well plates for experiments. To obtain complete differentiation, cells in cell culture dishes were maintained for 14 days before they were used in experiments.
Test concentrations
All substances were dissolved in DMSO and diluted in the respective medium to a final DMSO concentration of 0.5% before incubation. HepaRG treatment medium and 0.5% DMSO in RPTEC medium served as solvent controls for HepaRG cells and RPTEC, respectively. At least 3 biological replicates, i.e., independent experiments, were performed for each assay.
Cell viability
Cell viability was investigated with the WST-1 assay (Immunservice, Hamburg, Germany), according to the manufacturer’s protocol and subsequent NR uptake assay according to Repetto et al. (2008). HepaRG cells and RPTEC were seeded in 96-well plates and incubated with the test substances for 72 h. Triton X-100 (0.01%, Thermo Fisher Scientific, Darmstadt, Germany) was used as positive control for reduced cell viability. At the end of the incubation period, 10 µL WST-1 solution was added to each well and incubated for 30 min at 37 °C. The tetrazolium salt WST-1 is metabolized by cellular mitochondrial dehydrogenases of living cells to a formazan derivative, the absorbance of which was measured at 450 nm with an Infinite M200 PRO plate reader (Tecan, Maennedorf, Switzerland). The reading of each well was related to the absorbance value at the reference wavelength of 620 nm, and blank values were subtracted before the relation to the solvent control.
Afterwards the NR uptake assay was performed, where incorporation of NR into lysosomes of viable cells is measured. One day prior to the assay, NR medium was prepared by diluting a 4 mg mL−1 NR stock solution in PBS 1:100 with the respective cell culture medium for HepaRG cells and RPTEC, and incubated at 37 °C over night. After the WST-1 measurement, the incubation medium was removed and cells were washed twice with PBS. Subsequently, 100 µL NR medium, previously centrifuged for 10 min at 600 × g, was added and incubated for 2 h. Afterwards, cells were washed twice with PBS, and 100 µL destaining solution (49.5:49.5:1 ethanol absolute, distilled water, glacial acetic acid) per well was added. Plates were shaken at 500 rotations min−1 for 10 min and fluorescence of NR was measured with an Infinite M200 PRO plate reader (Tecan, Maennedorf, Switzerland) at 530 nm excitation and 645 nm emission. Each reading was subtracted by the blank value and normalized to the solvent control.
Multiplexed microsphere-based sandwich immunoassays
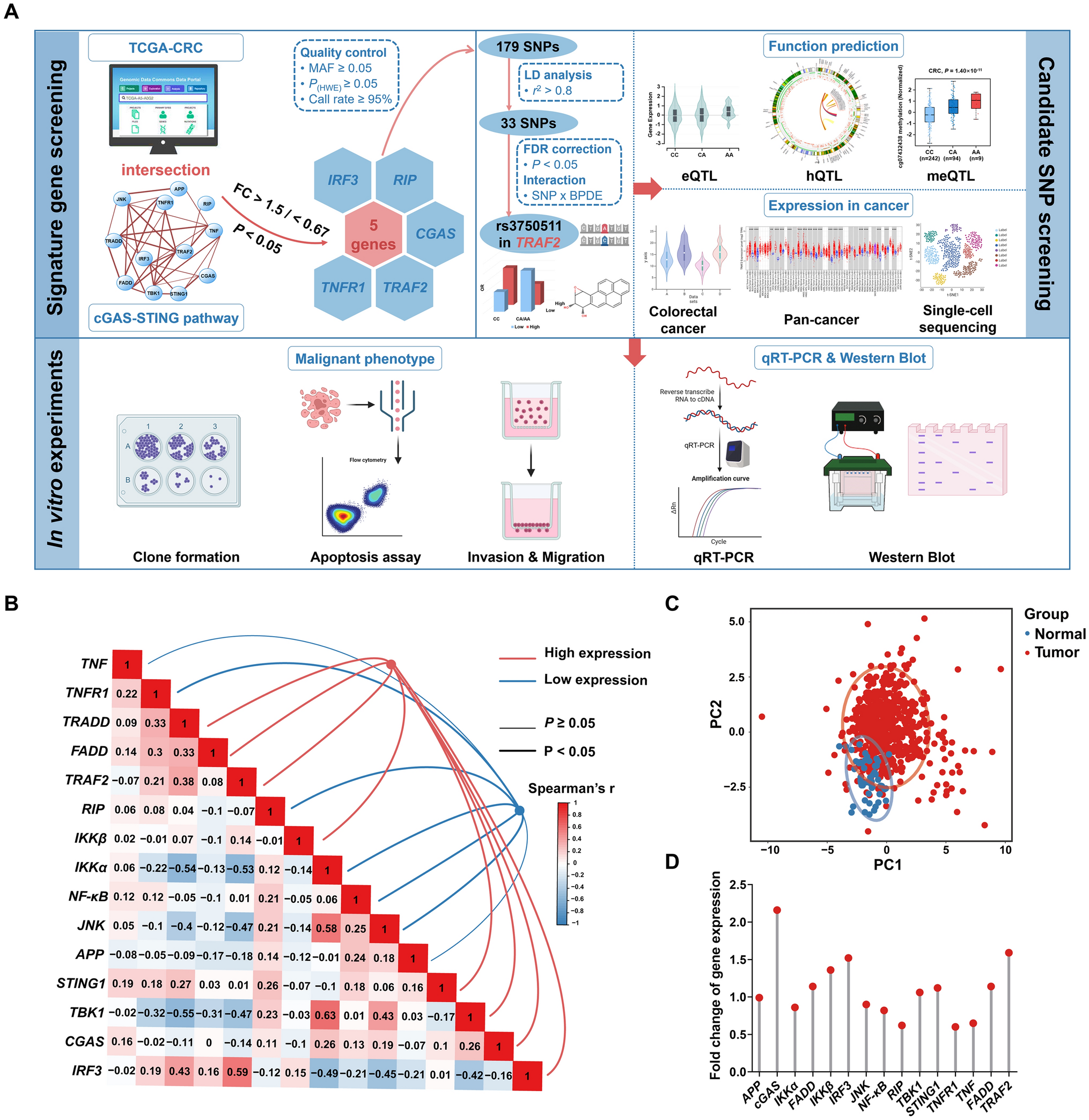
Marker proteins and protein modifications were analyzed by Signatope GmbH (Tübingen, Germany) with a multiplexed microsphere-based sandwich immunoassay. Cells were seeded in 6-well plates and incubated with the test substances for 36 and 72 h. Protein extraction was performed by adding 250 µL pre-cooled extraction buffer, supplied by the company, to the cells in each well and subsequent incubation for 30 min at 4 °C. Cell lysates were transferred to 1.5 mL reaction tubes and centrifuged for 30 min at 4 °C and 15 000 × g. The supernatant was aliquoted in 60 µL batches and stored at -80 °C until shipment. After thawing, aliquots were directly used and not frozen again. Samples were analyzed for 8 proteins and protein modifications, each representing a marker for a certain form of toxicity (Table 2).
Table 2 Analyzed proteins or protein modifications and associated cellular functionsQuantitative real-time PCR and PCR profiler arrays
RT-qPCR was conducted to ensure well performing RNA for subsequent PCR profiler arrays. Cells were seeded in 12-well plates and incubated with the test substances for 36 h. RNA extraction was performed with the RNA easy Mini Kit (Qiagen, Venlo, Netherlands) according to the manufacturer’s manual. Yield RNA concentration and purity were analyzed with a Nanodrop spectrometer (NanoDrop 2000, Thermo Fischer Scientific, Darmstadt, Germany) and RNA samples were stored at -80 °C until further use. Reverse transcription to cDNA was conducted using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Waltham, MA, USA) according to the manufacturer’s protocol with a GeneAmp® PCR System 9700 (Applied Biosystems, Darmstadt, Germany) and cDNA samples were stored at – 20 °C. RT-qPCR was performed with Maxima SYBR Green/ROX Master Mix (Thermo Fisher Scientific, Darmstadt, Germany) according to manufacturer’s protocol. In brief, 9 µL master mix, consisting of 5 µL Maxima SYBR Green/ROX qPCR Master Mix, 0.6 µL each of forward and reverse primers (2.5 µM) and 2.8 µL nuclease-free water, was added to each well of a 384-well plate. Primer sequences are shown in Online Resource 1. Subsequently, 20 ng cDNA was added to each well to a final volume of 10 µL and RT-qPCR was performed with an ABI 7900HT Fast Real-Time PCR system instrument (Applied Biosystems, Darmstadt, Germany). In brief, activation took place at 95 °C for 15 min, followed by 40 cycles of 15 s at 95 °C and 60 s at 60 °C, followed by 15 min at 60 °C and default melting curve analysis. Data were processed using 7900 software v241 and Microsoft Excel 2021. Threshold cycle (CT) was set to 0.5, melting curve was checked and manual baseline correction was performed for each gene individually. Yield CT-values were extracted to Microsoft Excel 2021 and relative gene expression was obtained with the 2−ΔΔCt method according to Livak and Schmittgen (2001). GUSB and HPRT1 served as endogenous control genes for HepaRG cells, GUSB and GAPDH were used for RPTEC. Primer efficiency was tested beforehand according to Schmittgen and Livak (2008). Only RNA samples showing amplification in RT-qPCR were used for further analysis with PCR profiler arrays. For quality control purposes, yield 2−ΔΔCt values from RT-qPCR and PCR profiler arrays were compared and had to be within the same range (Online Resource 1).
For performing the PCR profiler array, cDNA was synthesized from 1 µg RNA using the RT2 First Strand Kit (Qiagen, Venlo, Netherlands) according to the manufacturer’s protocol with a GeneAmp® PCR System 9700 (Applied Biosystems, Darmstadt, Germany). Subsequently, the RT2 Profiler™ PCR Array Human Molecular Toxicology Pathway Finder or Nephrotoxicity (Qiagen, Venlo, Netherlands) was conducted with RT2 SYBR® Green ROX qPCR Mastermix (Qiagen, Venlo, Netherlands) according to the manufacturer’s protocol. RT-qPCR was performed with an ABI 7900HT Fast Real-Time PCR system instrument (Applied Biosystems, Darmstadt, Germany), where activation of polymerase took place for 10 min at 95 °C, followed by 40 cycles of 15 s at 95 °C and 60 s at 60 °C and default melting curve analysis. Data were analyzed using 7900 software v241 and Excel 2021. CT was set to 0.2, melting curve was checked and manual baseline correction was performed. Yield CT-values were extracted and further analyzed.
Pathway analysis
Further evaluation of PCR array data was performed with functional class scoring methods such as Gene Ontology (GO) enrichment and Kyoto Encyclopedia of Genes and Genomes (KEGG), as well as with the bioinformatics analysis and search tool Ingenuity Pathway Analysis Software (IPA). Following the manufacturer’s instructions, yield CT-values were uploaded to the Qiagen Gene Globe WebportalFootnote 1 and analyzed using the standard ΔΔCT method referring to an untreated control. A cut-off CT was set to 35, all 5 built-in housekeeping genes were manually selected as reference genes and their arithmetic mean used for normalization. Means of fold regulation and p-values were calculated and further evaluated with the bioinformatics tools following the protocol provided in Online Resource 2. The processed results from HepaRG cells and RPTEC were used as input data individually, as well as combined. For the combined analysis, duplicate genes that were present on both arrays were removed.
To generate a first overview, the percentage of differentially expressed genes (DEG) per pathway was determined as previously published (Heise et al. 2018). Genes were assorted to pathways as suggested on the manufacturer’s web page.Footnote 2 The percentage of DEG was calculated as number of genes whose expression significantly differed by a fold change of 2, as determined by Student’s t-test (p < 0.05), related to the total number of genes in the pathway.
GO enrichment and KEGG analysis
The freely available web tools GOrillaFootnote 3 and ShinyGO 0.80Footnote 4 were used for GO enrichment and KEGG analysis, respectively (Eden et al. 2007, 2009; Ge et al. 2020). Detailed protocols are provided in Online Resource 2 together with the R code for determining DEG and background genes (see Data availability), which was adapted from Feiertag et al. (2023).
Ingenuity pathway analysis
In addition to GO enrichment and KEGG analysis, further evaluation of PCR array data was performed with the bioinformatics analysis and search tool IPA (Qiagen, Hilden, Germany, analysis date: Nov. 2023) as previously published (Karaca et al. 2023b). IPA is a commercial bioinformatics tool for analyzing RNA data, predicting pathway activation and functional interrelations using a curated pathway database. Using Fisher’s exact test, IPA identifies overrepresented pathways by measuring significant overlaps between user-provided gene lists and predefined gene sets. Means of fold regulation and p-values were uploaded to IPA following the protocol provided in Online Resource 2. Cut-off was set to – 1.5 and + 1.5 for fold regulation and 0.05 for the p-value. Fold regulation represents fold change results in a biologically meaningful way. In case the fold change is greater than 1, the fold regulation is equal to the fold change. For fold change values less than 1, the fold regulation is the negative inverse of the fold change. No further filtering was applied and an IPA core analysis was run. One Excel spread sheet per substance was obtained including all predicted diseases or functions annotations, the associated categories, the p-value of overlap as well as the number and names of the DEG found in the respective annotation (Online Resource 3). Predicted effects on other organs than the liver or the kidneys, such as heart or lungs, were discarded. For further comparison with in vivo data only the categories were used, combined with the p-value of the annotation, which was the highest.
Comparison with animal studies
The data obtained from targeted protein and transcriptomics analyses were compared with known in vivo observations from Draft Assessment Reports (DARs) of the pesticide active substances required for pesticide legislation. To facilitate the comparison of the data, the in vitro data was transformed into a more comprehensible form by applying evaluation matrices as shown in Table 3.
Table 3 Evaluation matrices for analysis of targeted protein and transcriptomics further analyzed with Ingenuity Pathway Analysis software (IPA)The in vivo effects attributed to the pesticide active substances were taken from the publication by Nielsen et al. (2012). Additionally, the DARs of the two substances not reported in Nielsen et al. were analyzed and assigned accordingly. All in vivo effects identified by the authors for liver and kidneys can be found in Online Resource 1. Based on expert knowledge, descriptions of in vitro outcomes were combined with in vivo observations (see Tables 4 and 5).
Table 4 Combination of prediction from marker proteins with potential in vivo observations as categorized by Nielsen et al. (2012)Table 5 Combination of prediction from Ingenuity Pathway Analysis (IPA) of transcriptomics data with potential in vivo observations as categorized by Nielsen et al. (2012)Based on the combination of the in vitro and the in vivo data, it was possible to draw conclusions on the concordance of the predictions. In order to establish optimized thresholds for regarding an effect as in vitro positive, the analyses were performed by considering at least medium effects, strong and very strong effects, or very strong effects only (see Table 3) and comparing these to the corresponding in vivo effect. In case multiple in vitro predictors were connected to the same in vivo observation, a positive prediction from one was sufficient to be considered in vitro positive. For protein analyses, the comparison was performed for the data from HepaRG cells and RPTEC individually, as well as combined, where a positive prediction from one of the cell lines was considered sufficient and compared to hepatotoxic and nephrotoxic in vivo effects. For the gene transcription analysis, the categories obtained by IPA were compared to in vivo observations from DARs. A further evaluation integrating protein and transcriptional data was conducted, wherein a positive result from either data type was sufficient to classify a sample as in vitro positive. Online Resource 1 shows the combination of the results in detail. The percentage of concordance between in vitro prediction and in vivo observation was calculated. Indicative concordance was defined as percentage of in vivo positive observations that were predicted to be positive by the in vitro test system.
Statistical analysis
Statistical analysis was performed using R 4.2.1 and RStudio 2023.09.1 + 494. Data evaluation was done with Microsoft Excel 2021.
All experiments were performed in at least three independent biological replicates. Technical replicates, when applicable, were averaged and subsequently mean and standard deviation values were calculated from biological replicates. For targeted protein analysis, statistical significance was calculated with bootstrap technique using R package boot (Canty and Ripley 2016; Davidson and Hinkley 1997) to account for the high variability that results when the protein expression is affected. Data visualization was done using ggplot2 package (Wickham 2016). Calculation of statistical significance of altered gene transcription was performed using Student’s t-test, and R package ComplexHeatmaps was used for data visualization (Gu 2022). All R scripts can be found using the link provided in the Data availability section.





留言 (0)